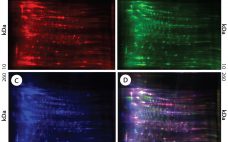
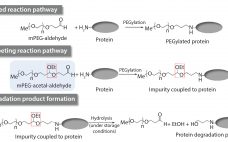
Exposure to solid–liquid and air–water interfaces during production, freezing and thawing, shipment and storage of protein therapeutics may be a contributing factor in their degradation (e.g., aggregation, fragmentation) (1, 2). Surface exposure, particularly during manufacturing processes, often is accompanied by various degrees and durations of shear stresses originating from fluid flow and acting on proteins at interfaces. The magnitude and duration of shear rates depends on velocity gradients within each solution and varies significantly among manufacturing steps. On the low…