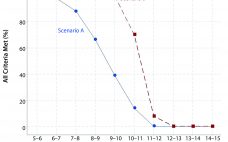
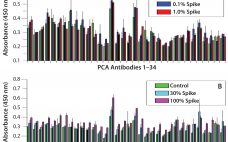
The primary goal of container–closure integrity (CCI) is to maintain the sterility and product quality of parenteral biopharmaceuticals throughout their shelf life and use. Guidelines detailing the initial CCI qualification and validation requirements have been defined and can be found in the US Pharmacopeia chapter 1207 (USP<1207>) (1). The guidelines described in USP<1207> can be applied to any common CCI testing (CCIT) method to achieve a method suited for its intended use within a drug product lifecycle. CCI is not…