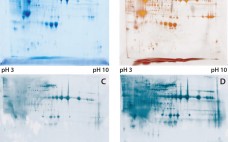
This is a transcript from a Q&A interview with Emily Shacter, PhD, Consultant, ThinkFDA LLC (former FDA Scientist and Regulator). We will be talking today about the CMC Forum that was published back in 2005. We are revisiting it in the magazine to specifically update our understanding of how to maintain process control; understanding your process. In general, how do you feel the discussions in the four-part paper from 2005 has held up after 10 years? Emily: I think they…