Regardless of the industry and product being manufactured, continuous processing has demonstrated numerous benefits. In addition to smaller manufacturing footprints, reduced material consumption and waste generation, increased efficiencies, and lower capital and operating costs continuous manufacturing typically leads to more consistent processes and product quality. In the pharmaceutical industry, the latter two attributes align perfectly with FDA’s Quality by Design (QbD) and process analytical technology (PAT) initiatives. The challenge is determining how to apply these concepts in practice. Applying the…
Regulatory Affairs
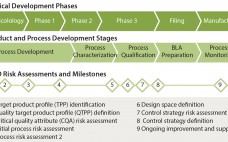
Quality By Design for Monoclonal Antibodies, Part 1: Establishing the Foundations for Process Development
The quality by design (QbD) modernized approach to pharmaceutical development is intended to provide regulatory flexibility, increased development and manufacturing efficiency, and greater room to innovate as well as improve manufacturing processes within defined ranges without obtaining regulatory approval first. QbD is a systematic developmental approach that starts with a clear goal in mind and emphasizes understanding of how variability in both process and materials affects a final product (1). Historically, product quality has been assured either with end-product testing…
A Turning Point for US Biosimilars
The next 12–18 months could be a critical time for biosimilars in the United States (1). This product class has grown rapidly since passage of the Biosimilars Price Competition and Innovation Act (BPCIA) of 2010. That landmark legislation allowed for biosimilar market approvals based on previously approved “reference products,” creating an expedited pathway that reduces biosimilar development costs and speeds regulatory review so patients get faster access to essential medicines. Increased competition from biosimilars could save the US healthcare system…
Automation of CAR-T Cell Adoptive Immunotherapy Bioprocessing: Technology Opportunities to Debottleneck Manufacturing
Continued clinical efficacy demonstrations of cell-based immunotherapies (iTx) such as chimeric antigen receptor T cell (CAR-T) therapies has made the prospect increasingly likely of an immunotherapy product achieving conditional market authorization in the short term. For example, Novartis and the University of Pennsylvania’s lead candidate (CTL019) for treating a range of hematological malignancies received breakthrough status from the US Food and Drug Administration (FDA) in 2014, permitting access to an expedited drug development pathway for high unmet medical needs (1).…

CMC Strategy Forum Special Focus Series: Part 2 Product-Related Impurities, An Overview
Introduction by Cheryl Scott The CMC Strategy Forums focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the life cycle of a therapeutic and thereby foster collaborative technical and regulatory interaction. Forum chairs share information with regulatory agencies to help them merge good scientific and regulatory practices. Outcomes of forum meetings are published in BioProcess International and on the CASSS website. This process is meant to help ensure that biopharmaceutical products manufactured with advancing technologies in a regulated environment…
Addressing Variability in Product Labeling: Explore Dynamic Labeling for Your Global Enterprise
Adding to the complexity of drug-product labeling, companies face a broad range of evolving requirements. Those include regional, language, customer, and regulatory requirements that must be met quickly and efficiently to prevent supply-chain disruption. Companies that cannot meet those requirements can end up with fines, dissatisfied customers, and loss of business. Enterprise labeling solutions allow drug makers to deal with variability in labeling by providing label formatting that supports myriad different label combinations with a minimum number of label designs.…

The Year of Data Integrity: 2015 Brought a Worldwide Focus on Training, System Design and Control, and Data Management
Each year, regulatory agencies from around the world focus on critical aspects of the pharmaceutical quality management system, bringing awareness to the industry and continuing to effect positive change. In the past five years, risk assessments, electronic records, and outsourced activities have been in the spotlight. As 2015 closed out, it was clearly the year of data integrity. In March 2015, the UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) published its GMP Data Integrity Definitions and Guidance for Industry,…

Defining Your Product Profile and Maintaining Control Over It | A Look Back with Emily Shacter
This is a transcript from a Q&A interview with Emily Shacter, PhD, Consultant, ThinkFDA LLC (former FDA Scientist and Regulator). We will be talking today about the CMC Forum that was published back in 2005. We are revisiting it in the magazine to specifically update our understanding of how to maintain process control; understanding your process. In general, how do you feel the discussions in the four-part paper from 2005 has held up after 10 years? Emily: I think they…

Responding to an FDA Form 483: A Five-Step Approach
When the US Food and Drug Administration (FDA) inspects your company’s biomanufacturing facility, investigators use the FDA Form 483 to record observations and findings (1). Such inspections typically review all good manufacturing practices and good laboratory practices (GxP) quality systems documents. If those investigators find compliance issues, they deliver a summary of their observations and findings using a Form 483, a copy of which will be provided to your company at the end of the inspection visit. How to Respond…
Bridging Analytical Methods for Release and Stability Testing: Technical, Quality and Regulatory Considerations
To monitor the control and consistency of products derived from biological systems, a broad array of analytical methods are used for biopharmaceutical release and stability testing. These methods include both classical and state-of-the-art technologies as well as new technologies as they emerge over time.During the life cycle of a product, several reasons can arise for making changes in existing analytical methods: e.g., improved sensitivity, specificity, or accuracy; increased operational robustness; streamlined workflows; shortened testing times; and lowered cost of testing.…