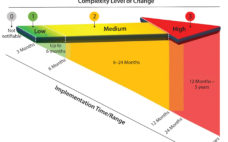
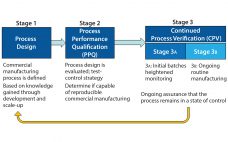
Opportunities for establishing strong biopharmaceutical capabilities are expanding across the globe. This e-book seeks to encapsulate the current state of emerging markets/countries, tracing key elements above and offering examples to show where (in the world) the biopharmaceutical industry is expanding and securing its footholds. Generally, to succeed in these markets, foreign companies must exercise efficient resource management and control, show creativity and receptiveness to cultural differences, develop new strategies, and manage expectations. Working with local partners can provide access to…