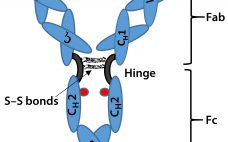
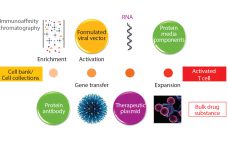
The BioPhorum Operation Group’s (BPOG’s) Container Closure Integrity Testing (CCIT) workstream would like to congratulate the United States Pharmacopeia’s committee for its latest revision to USP chapter <1207> Package Integrity Evaluation: Sterile Products. Generally, we believe it provides a comprehensive overview of the available methods for container–closure testing and outlines many important elements for consideration in establishing a successful CCIT strategy. We first responded to the USP <1207> draft when it was released for comment in 2014. And from our…