 As anyone who has focused on host-cell proteins as process contaminants can tell you, trying to purify a specific type of molecule from a large mixture of many similar molecules is like trying to find a few particular needles in a huge pile of varied needles. The same could be said for purifying viral vectors from cell culture fluids. When viruses are the products, unwanted viruses are contaminants that must be separated away — or better yet, prevented from being there in the first place.
As anyone who has focused on host-cell proteins as process contaminants can tell you, trying to purify a specific type of molecule from a large mixture of many similar molecules is like trying to find a few particular needles in a huge pile of varied needles. The same could be said for purifying viral vectors from cell culture fluids. When viruses are the products, unwanted viruses are contaminants that must be separated away — or better yet, prevented from being there in the first place.
Viral safety is a critical aspect of downstream processing for biologics, especially those derived from mammalian cell lines. And it’s not only about protecting patients from infection; it’s also about protecting the drugs from unwanted immune responses. Such responses dampened early hopes for adenoviruses as gene-therapy vectors — some serotypes are so common in the environment that many patients are already primed and readied to respond to their presence with an attack. To make things worse, some people’s bodies can overreact with catastrophic results (1).
These days, the most promising and popular candidates for gene-therapy vectors are adenoassociated and lentivirus species — both relatively small as viruses go (Figure 1). Being small makes them more of a downstream processing challenge in general; it’s a lot easier to catch bacteria and big viruses with a filter that lets proteins and smaller viruses move through, but some adventitious agents of concern are relatively small, too. Human immunodeficiency virus (HIV) is a lentivirus, after all, and in fact one of those in development as a vector (safeguarded and “defanged,” of course). And parvoviruses are not only about half the size of HIV, but they also are linear in structure, giving them the potential to squeeze through even the tight pores of nanofilters. So process engineers must exploit other physicochemical properties of their products to prevent parvovirus contamination. Again, it’s better to prevent that from ever becoming a concern, but you also need a process with a demonstrated ability to clear adventitious agents just in case.
So viral safety isn’t just about viral clearance (removal). It also includes inactivation of such contaminants — “killing them” or making them unable to replicate — and crucially, keeping them away from the start. You can’t do any of those things without good detection methods to use in process development and to confirm your results. This is the three-pronged approach recommended by regulators around the world (2–4):
- Prevent contamination of raw materials
- Detect potential contaminants through characterization studies and regular testing
- Eliminate contaminants (either inactivation or removal).
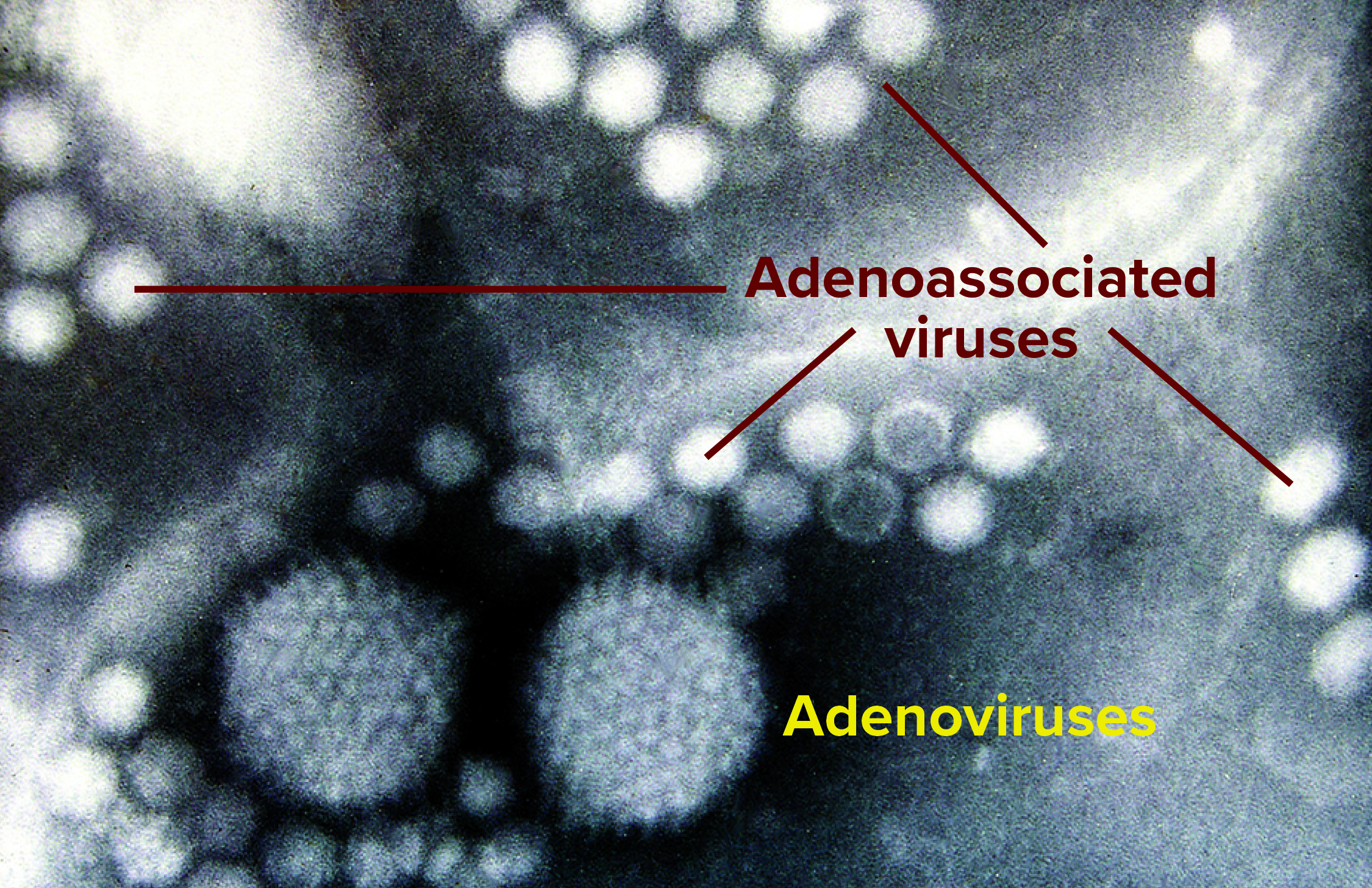
Figure 1: Two adenovirus particles surrounded by numerous smaller adenoassociated viruses magnified at ~200,000× (Graham Beards, Wikimedia Commons, https://commons.wikimedia.org)
Prevention Paramount
If any animal-sourced materials are used in cell culture media for biopharmaceutical production, they must be qualified for good manufacturing practice (GMP) use and virus-filtered (by the supplier or user, or both). This is less of a concern in bioprocessing of antibodies and other proteins, for which manufacturers increasingly have turned toward animal-free sources of media components in recent years. That trend was driven early on by concern over transmissible spongiform encephalopathies (TSEs) from prion proteins — which are even smaller than viruses and harder to get rid of. But over the years, it’s had a positive impact on virus safety as well.
However, many gene and cell therapy processes come out of academic environments, where animal-sourced ingredients are used more often. As those processes move into industrial settings, sometimes companies are so focused on scale-up/-out and technology transfer that viral safety can become an afterthought — although it absolutely should not. Some developers have neglected to use virus filters in their upstream processes because they expect downstream processing to handle such concerns. But they are important to decreasing bioburden when many media components are labile and cannot be subject to routinely used heat-inactivation methods. After the first Parenteral Drug Association (PDA) meeting on viral safety of advanced therapies in 2016, more attention has been given to the three-pronged approach for cell, gene, and tissue therapies.
“Prevention is even more key in viral vector and gene therapy applications than in traditional bioprocessing,” says BPI editorial advisor Hazel Aranha (GAEA Resources). “Considerations are similar in both, with an emphasis on raw-material sourcing, documentation, and avoidance of animal-derived materials (ADMs).”
BPI’s June 2021 featured report highlighted seldom-considered challenges related to cell banking (5–7). We collected impressions from biopharmaceutical developers, contract manufacturing organizations, and other industry partners about best practices for establishing, transferring, handling, and storing research, master, and working cell banks (RCBs, MCBs, WCBs). I followed up with Aranha to focus on such considerations for viral vector bioprocesses. A PhD microbiologist, she is regulatory affairs certified (RAC) for both the United States and the European Union, with over three decades of experience in academia, industry, and consulting. She has worked for Catalent Pharma Solutions, Pall Corporation, Sartorius Stedim Biotech, and Wyeth Vaccines, all primarily in virus safety. With her extensive knowledge of virology, she’s turned her interest in recent years to gene therapies — in particular those using viral vectors.
To make advanced therapy medicinal products (ATMPs) commercially viable, she explained, it would be ideal to establish MCBs and WCBs, as with other biopharmaceuticals. “However, we are not there yet,” Aranha says. Some companies are working on such an approach for gene therapy processes based on infection of mammalian cell cultures, but early stages in propagation still require growth in serum-supplemented media. Those cell lines will have to be weaned away from animal-derived nutrients and adapted to chemically defined media. “Many companies are still working toward that goal.”
For gene-modified cell therapies, the viral vector that delivers genetic material to donor cells is considered to be a critical raw material; otherwise, it’s a final therapeutic product for gene therapy delivered to patients without a cell-therapy component. Either way, strict attention must be paid to vector quality and associated documentation.
“Begin with the end in mind,” Aranha advises. “When the intention is to progress an operation into clinical applications and commercial manufacturing, it is important to work toward that goal. Use materials of appropriate grade and ensure best practices during establishment and maintenance of viral vector cell banks — and the cell lines used to produce them.”
Research groups must keep best practices in mind, she cautions, because cells used early in development might be needed later for unanticipated purposes. In addition to producing clinical study materials, it may be necessary to use those cells and tissues as reference material for data interpretation when demonstrating in vivo equivalence.
When a basic research protocol gets applied to clinical studies, the quality of reagents and starting materials must improve — from “for research only” to well-sourced materials with documented provenance. Often only research-grade materials are available for gene-therapy work; when that is the case, regulators recommend that the highest grade available be used with appropriate documentation provided. New ATMPs developed in academic institutions with research-grade reagents and untested starting materials cannot provide the minimum quality standards required for biomanufacturing.
In the past, Aranha recalls, research groups would outsource the production of the viral vectors they needed based on plasmids developed in house. Academics would leave the GMP work to a contract manufacturing organization (CMO) with expertise in regulatory compliance and an understanding of and capability to meet GMP requirements. Now, biopharmaceutical companies have acquired or established their own research groups for generating potential gene-therapy candidates. Those groups exert more control over the entire process than purely research organizations would.
“But keep in mind,” Aranha points out, “that GMPs are mandatory for all manufacturing sites dedicated to ATMP production, including those operated by academic institutions.” Key aspects include quality assurance and control (QA/QC), establishment and maintenance of expression systems with demonstrated understanding of critical quality attributes (CQAs), training through standard operating procedures (SOPs), risk management strategies, and up-to-date documentation of all the above.
Establishment and maintenance of the expression system includes cultivating a thorough understanding of key influencing factors: authenticity (identity, provenance, and confirmation of genotypic and/or phenotypic characteristics), purity (freedom from contamination, biological or otherwise), cell-line stability (genotypic and phenotypic) in culture and in cryostorage, and functional integrity of the in vitro system. Standardization of expression systems is at issue not only during their establishment, but also throughout their lifetime of subsequent manipulations, maintenance, cultivation, and preservation activities over years to come.
Assurance of quality for all materials and robustness of methods helps a company maintain the integrity, validity, and reproducibility of its product. Documentation of all information needed to track those materials and methods makes the work repeatable in the future and helps reviewers understand and evaluate it when investigational new drug (IND) and market licensing applications are filed.
Protection of individuals and the environment from potential hazards requires establishment and maintenance of adequate measures such as closed systems, barrier–isolators, cleanrooms, and set routes for air, material, and personnel flows. Compliance with relevant laws and regulations — and with ethical principles — is especially of concern with ATMPs. All of this requires provisions for relevant and adequate education and training of all personnel to promote both safety and high-quality work.
“When using a CMO,” Aranha says, “ensure that you have audited the facilities and have a quality agreement in place. Obtain basic information related to the process: materials quality, grade, and traceability, with certifications and statements including certificates of analysis (CoA) for batches used in development. Some CMOs have drug master files (DMFs) with the relevant authorities, and in such a case a letter of authorization (LoA) would suffice.”
Risk management is crucial, Aranha says, in evaluating every stage of a biomanufacturing process and evaluating needs for scale-up/-out procedures. “Risk assessment is implied in the United States; in Europe and many other parts in the world it is a requirement.”
Many researchers and companies use human embryonic kidney (HEK293) cells or Spodoptera frugiperda (Sf9) insect cells for producing adenoassociated viruses (AAVs), retroviruses, adenoviruses, or lentiviruses through transient transfection. Proponents of the baculovirus expression vector system (BEVS) tout the relative viral safety of insect cells over mammalian cells for producing biotherapeutics. Few if any endogenous or adventitious viruses can infect both insects and humans.
To guarantee product robustness and homogeneity, the cell line used for clinical/commercial applications must be analyzed to document its quality parameters and attributes. Authorities around the world have produced many regulations and guidance documents on production of GMP cell banks. For example, ICH Q5B provides detailed information on analyzing expression constructs and cell clones used to develop MCBs (8). ICH Q5D on derivation and characterization of cell lines states that their identity, purity, and stability (and that of subsequent cell banks) must be confirmed (9).
Even as the biopharmaceutical industry at large has moved away from using ADMs such as serum and specific enzymes, few viral vector production systems have been weaned away from serum. They commonly include fetal bovine serum (FBS) and porcine trypsin in media formulations for propagation of producer cells. Aranha cautions that even when ADMs are not used directly, supplements derived from them might be used. “That imposes the same cautions as with any other biotech products. Some companies use serum supplements when initially setting up a cell bank and then slowly wean the cells off those materials by decreasing their concentrations over time.”
Using FBS incurs the risk of adventitious bovine viruses and prions. Therefore, documentation must include the results of testing for bovine viruses and include a CoA showing that the serum does not come from countries where BSE is found. Porcine trypsin brings the risk of patient exposure to porcine circovirus (PCV). In one case with PCV contamination of a marketed rotavirus vaccine, new diagnostic methods facilitated its detection, so they can be used to test porcine-sourced materials that must be documented as negative for that contaminant. Aranha says that sufficiently documented producer cell lines might be usable without those documented results — but a user would have to pe/rform such testing in house (10). “Once your MCB is tested or shown to have an accepted history of nonexposure to these agents,” she explains, “then these tests may be omitted in subsequent stages of production if CoAs for FBS and porcine trypsin contain the proper testing and come from approved geographic locations.”
Clearly, testing and documentation of assay results are vital components of viral safety, no matter what type of biologic a company is developing. Key analytical methods when it comes to viral safety include those based on polymerase chain reaction (PCR), cell-based infectivity and immunoassays, in vivo animal testing, and transmission electron microscopy (TEM). Screening assays are chosen based on the source of the cell substrate. Cutting-edge, next-generation sequencing (NGS) is only beginning to be used for detection of viruses and other contaminants in parental and banked cell lines.
Downstream Dilemmas
As important as preventing viral contamination in upstream production is, a corresponding downstream process must be ready to handle the unexpected. “Of the tripronged approach to viral safety,” Aranha says, “removal is most challenging for viral vector products, whether the virus is the product of interest or a critical raw material. Gene-therapy products are relatively labile, so viral inactivation and removal methods typically used in bioprocessing cannot be applied here. Thus, control of raw materials and stringent application of aseptic methods are key to ensuring that a final product is free of microbial contaminants and other adventitious agents.
“In spite of these limitations,” she continues, “the industry has been successful in producing safe vaccines for years. Vaccine product purification methods that serendipitously provide virus inactivation can be validated. For example, virus vaccine processes have used detergent steps that can be validated, as can other purification methods such as ion-exchange chromatography. However, virus-removal filtration intended to remove small viruses cannot be used.”
Anything that would disrupt a contaminating virus also is likely to inactivate and damage viral vectors. From this standpoint, AAVs are less problematic than lentiviruses. Good yields are possible after detergent/pH treatments or virus removal filters with pore-size cutoffs for viruses 50 nm or larger. Such steps would irreparably damage or remove lentiviruses. Although no reports have arisen yet of contamination during manufacture of viral vectors, Aranha points out, one blood collection site did report bacterial contamination that was traced to a handling issue.
Last winter, BPI’s managing editor Maribel Rios reported on discussions of viral vector processing during Informa’s virtual Cell and Gene Bioprocessing and Commercialization Conference and Exhibition (11). She highlighted a panel discussion with Jayanthi Grebin (Colder Products Company), Thomas Krell (Takeda) and and Sam Wadsworth (Ultragenyx), at which viral safety came up during the question-and-answer session. Panelists were asked to comment on the main challenges in when validating a viral clearance filter panel for potential sources of adventitious viruses — and to compare with those for monoclonal antibodies (MAbs). “For different gene therapy vectors,” Kreil said, “an upstream viral barrier is entirely doable.” He pointed out that the volumes that must be handled are smaller for viral vector products than for MAbs. “There are midsize nanofilters that allow for the passage of these vectors and still retain adventitious viruses.”
But viral clearance for lentiviral vectors could be more difficult than for other viral vectors. Grebin pointed out that lentiviruses (associated with ex vivo therapies) are more challenging to purify because they are bigger than in vivo viruses such as AAV. So a 0.2-µm filter might not be usable with lentivectors. “I think our hands are tied in the manner of what viruses we use. There are challenges in the viruses you use based on the type of treatment that you want to do for your process [in vivo or ex vivo].” Kreil added, “There is a complication with every manipulation that needs to occur before a treatment is brought back to a patient. If you do the production right, you can use closed systems that [do not require] interference with operators. But with an ex vivo system, there is more exposure [and] more manipulation that also is going to provide more opportunities for contamination events.”
On a follow-up question regarding viral contamination precautions for cell and gene therapies, Kreil said, “We need to strike a careful balance in making those treatments available, and at the same time making sure they come within an adequate safety margin. Essentially, the technology is there; we have a toolbox to prevent [contamination].”
Wadsworth brought up that one common source of contamination is serum, which can be removed from a process as early as the cell-banking step. Single-use components also offer a major benefit. Grebin suggested that manufacturers evaluate their whole processes when it comes to preventing contamination, not just downstream steps. “What you’re doing upstream [to prevent contamination] is going to improve your process downstream. In some cases, you won’t have a good method for filtering impurities and contamination, so the upstream platform you use and how well you are getting your validation done will affect contamination.”
A holistic approach to measuring the virus-removal capabilities of a downstream process is common in the biopharmaceutical industry, where developers measure and report the log-reduction value (LRV) of each step in separation and purification in challenge studies with model viruses, then determine the cumulative virus clearance provided by those purification steps. For viral vectors, the downstream platform typically includes one or more column-chromatography or membrane-adsorber steps (12). Hydrophobic-interaction chromatography (HIC) could work for lipid-enveloped viruses; gel-filtration or size-exclusion chromatography (SEC) separates solution components by size; and ion-exchange chromatography (IEC) exploits electrostatic interactions between matrix-bound ligand/receptor molecules with viral capsids. The specific choices made depend on the physicochemical properties of the viral vector itself. Because adenoviruses and lentiviruses are negatively charged at neutral pH, for example, anion-exchange adsorbents can be used to purify them. Heparin-affinity chromatography can be used to capture both AAVs and lentiviruses — but heparin itself is typically animal derived, potentially bringing an unwanted risk of contamination into the downstream process.
Promise and Provisos
Aranha reports that the global viral vector manufacturing market is projected to grow at a compound annual growth rate (CAGR) of 22.09% to reach a market size of US$1,469.144 million in 2025 (up from $443.592 million in 2019). “The cancer subsegment is expected to be the fastest growing disease segment.”
Viral vector technologies — mostly AAV, lentivirus, and retroviral vectors — are in development for vaccines and therapeutics to treat both infectious and noninfectious diseases. Interest in viral vectors is on the rise, especially for use in vaccine development. Despite early troubles (and with technological improvements since), adenoviruses are established as a vaccine platform, with approvals in the past year for both Ebola and COVID-19.
For cancer therapy, viral vectors can be programmed to replicate specifically inside cancer cells and cause toxic effects that lead to apoptosis. Aranha says that an attractive feature of viral vectors is their capability to provide high levels of transgene expression in a broad range of host cells. “They are widely seen as means of meeting the United Nations’ sustainable development goals (SDGs) for 2030.”
In the United States, regulatory encouragement and patient advocacy groups have pushed rare-disease clinical research to the center stage. Orphan-drug incentives and other fast-track approaches have encouraged pharmaceutical and biotechnology companies to consider the development of rare disease medicines as a potentially-profitable venture. Here, too, viral vectored gene therapies are making inroads.
With all those expanding applications, it’s important for developers to start with the end in mind: quality, efficacy, and safety of viral vector products. The more patients are served by this modality, the more potential there is for exposure and dangerous outcomes. Preventing contamination with unwanted viruses — and using trustworthy processes that can catch and eliminate those should a breakthrough occur — is vital to the future success of these treatments, their manufacturers, and stakeholders across the biopharmaceutical ecosystem.
References
1 Rinde M. The Death of Jesse Gelsinger, 20 Years Later. Distillations 4 June 2019; https://www.sciencehistory.org/distillations/the-death-of-jesse-gelsinger-20-years-later.
2 CBER. Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications: Guidance for Industry. US Food and Drug Administration: Rockville, MD, February 2010; https://www.fda.gov/media/78428/download.
3 Technical Report 978, Annex 3. Recommendations for the Evaluation of Animal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks. World Health Organization: Geneva, Switzerland, 2013; https://www.who.int/biologicals/vaccines/TRS_978_Annex_3.pdf.
4 5.2.3: Cell Substrates for the Production of Vaccines for Human Use. European Pharmacopoeia: Strasbourg, France, 2018; https://www.scribd.com/document/429298608/5-2-3-Cell-Substrates-for-the-Production-of-Vaccines-for-Human-Use.
5 Gazaille B, Castillo F, Nims RW. Cell Banking in the Spotlight: Advising Biologics Developers About Cell Bank Preparation and Characterization. BioProcess Int. 19(6) 2021: S1–S4, S13; https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2021/06/19-6-FR-Castillo-and-Nims.pdf.
6 Montgomery SA, Wheelwright SW, Poon HF. Contractor Perspectives: Best Practices for Transfer, Handling, Testing, and Storage of Cell Banks. BioProcess Int. 19(6) 2021: S5–S9; https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2021/06/19-6-FR-CB-CDMO.pdf.
7 Ikonomou L, et al. Cell Banking for Cell and Gene Therapy: Regulatory, Scientific, and Ethical Considerations. BioProcess Int. 19(6) 2021: S10–S13; https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2021/06/19-6-FR-ISCT-reprint.pdf.
8 ICH Q5B: Analysis of the Expression Construct in Cells Used for Production of r-DNA Derived Protein Products. US Fed Reg. 61, 1 February 1996: 7006; https://database.ich.org/sites/default/files/Q5B%20Guideline.pdf.
9 ICH Q5D: Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products. US Fed Reg. 63(182)1998: 50244–50249; https://database.ich.org/sites/default/files/Q5D%20Guideline.pdf.
10 CFR Part 113.47. Detection of Extraneous Viruses By the Fluorescent Antibody Technique. US Fed Reg. 60, 9 May 1995: 24548; https://www.gpo.gov/fdsys/pkg/CFR-2001-title9-vol1/pdf/CFR-2001-title9-vol1-sec113-47.pdf.
11 Rios M. Optimization of Processes and Advanced Platforms for Viral Vector Processing. BioProcess Int.18(11–12) 2020: S1–S4; https://lne.e92.mwp.accessdomain.com/manufacturing/cell-therapies/optimization-of-processes-and-advanced-platforms-for-viral-vector-production.
12 Fuerstenau-Sharp M, et al. Scalable Purification of Viral Vectors for Gene Therapy: An Appraisal of Downstream Processing Approaches. BioProcess Int. 15(2) 2017: S12–S17; https://lne.e92.mwp.accessdomain.com/downstream-processing/separation-purification/viral-vector-purification-gene-therapy-appraisal-downstream-processing.
| Further Reading |
| As endorsed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) assembly in June 2019, a Q5A(R2) working group is revising the Q5A(R1) guideline “Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin.” This revision will consider new biotechnology product modalities, biomanufacturing technology advances, new analytical methods for virus testing, and scientific knowledge that has accumulated since publication of the original document in 1999. See https://ich.org/page/quality-guidelines for more information.
There are no specific viral vector regulations yet. Several guidelines were issued in 2020–2021, none specific to viral vectors. Here’s the link to US Food and Drug Administration’s (FDAs) page for help in developing gene therapies: https://www.fda.gov/vaccines-blood-biologics/biologics-guidances/cellular-gene-therapy-guidances. Apte A, et al. Manufacture and Regulation of Cell, Gene, and Tissue Therapies, Part 1: Chemistry, Manufacturing, and Control Challenges. BioProcess Int. 18(11–12) 2020: S6–S12; https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2020/12/18-11-12-FR-Apte.pdf. Cameau E. Overcoming Obstacles in AAV Viral Vector Manufacturing. BioProcess Int. 19(6) 2021: 68–69; https://lne.e92.mwp.accessdomain.com/sponsored-content/overcoming-obstacles-in-aav-vector-manufacturing. Krakowiak J, Liu Q, Nascimento-Brooks L. Rapid Development of Viral Vector Production Processes: Iterative Parameter Optimization. BioProcess Int. 19(4) 2021: 48–49; https://lne.e92.mwp.accessdomain.com/sponsored-content/modde-software-for-rapid-development-of-viral-vector-production-processes-iterative-parameter-optimization. Mathot F, Bourles E. Development of a Freeze-Dried Ebola-Expressing Adenoviral Vector: Unexpected Findings and Problems Solved. BioProcess Int. 16(5) 2018: 26–31; https://lne.e92.mwp.accessdomain.com/manufacturing/formulation/development-of-a-freeze-dried-ebola-expressing-adenoviral-vector-unexpected-findings-and-problems-solved. Mora A, Ezzyat Y. Anticipating Cell-Line Challenges to Drive CMC Readiness. BioProcess Int. 18(6) 2020: 52–58; https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2020/06/18-6-Mora.pdf. Snyder MA. Scalable Purification Strategies for Viral Vectors and Vaccines. BioProcess Int. webcast 26 September 2019: https://lne.e92.mwp.accessdomain.com/sponsored-content/scalable-purification-strategies-for-viral-vectors-and-vaccines. |
Cheryl Scott is cofounder and senior technical editor of BioProcess International, part of Informa Connect; 1-212-600-3429; cheryl.scott@informa.com.