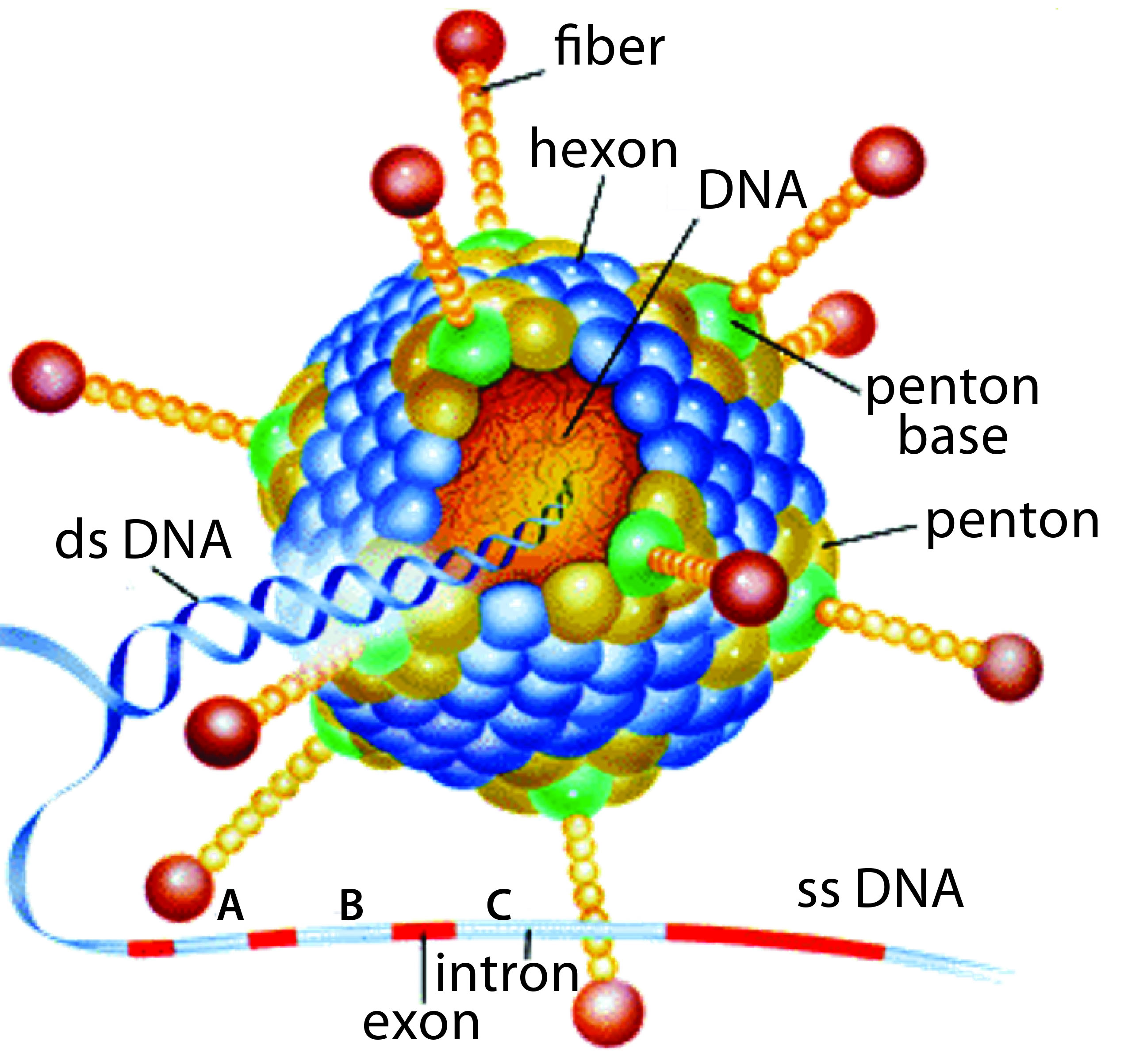
Replication-defective (E1 gene-deleted) chimpanzee type-3 adenovirus (ChAd3) (HTTPS://MICROBEWIKI.KENYON.EDU/INDEX.PHP/ ADENOVIRUS-BASED_GENE_THERAPY:_A_PROMISING_ NOVEL_CANCER_THERAPY)
In December 2013, a two-year-old child in Guinea became the first person to be killed by Ebola in the most recent outbreak. In March of the following year, that outbreak was declared in West Africa. By mid-2014, the World Health Organization (WHO) had declared it to be a public health emergency of international concern and urged pharmaceutical companies to accelerate their development of candidate vaccines. At the peak of the outbreak in 2014, more than 1,200 new cases of Ebola were reported every week in Liberia, Sierra Leone, and Guinea. This was the most significant outbreak in Africa during the past 50 years.
In acknowledgment of WHO’s request, GSK initiated an Ebola project and gathered staff with relevant expertise to push vaccine candidates from laboratory scale to a large-scale product. The selected product candidate was developed by the Okairos Company (now Reithera), a small biotechnology company from which GSK acquired its technical platform just one year before the Ebola outbreak. Okairos had developed an innovative technology based on adenoviruses isolated from different species of primates, primarily chimpanzees. Previously, the company had collaborated with the US National Institutes of Health (NIH) to develop its adenoviral vector: a replication-defective (E1 gene-deleted) chimpanzee type-3 adenovirus (ChAd3, pictured) carrying a transgene encoding the glycoprotein of Zaïre Ebolavirus (1).
Using chimpanzee adenoviruses brings two main benefits. First, adenoviruses naturally elicit a CD8+ T-cell response, which is essential to fighting intracellular pathogens. Second, humans have low preexisting immunity against chimpanzee vectors — high preexisting immunity being a major drawback with using human-specific viruses.
To produce these adenoviruses, GSK uses a complementing cell line (HEK 293) expressing human Adenovirus 5 E1 sequence, allowing the growth of replication-defective, E1-deleted ChAd3. When used as a vaccination, the resulting viruses can infect and use cell machinery to express the transgene (in this case the favored Ebola transgene), but they cannot replicate without their own E1 sequence (2). In fact, the transgene is located where the E1 sequence was deleted.
The adenovirus vector technology introduces a new way to vaccinate people. Indeed, the concept of vaccination has evolved has evolved over a century from an empirical approach using natural pathogens through recombinant technology to produce pure and well-defined antigens. Unfortunately, the latter have not proven to elicit high levels of immunity. Conjugation and adjuvants were developed as a response in the 1990s. And in 2010, a reverse-vaccination approach (sequencing the whole pathogen genome and isolating key antigens) was at the origin of a meningococcal vaccine (3). The next-generation products include glycoconjugates, self-amplifying messenger ribonucleic acid (mRNA), new adjuvants, and adenoviruses.
A Scale-Up Challenge
GSK faced multiple and concomitant challenges for transforming a laboratory-scale vaccine candidate into a commercial-scale product in just a few months (instead of years). GMP cell banks and virus seeds were needed for ChAd3-EBO-Z vaccine manufacturing, and production yield had to be increased by 1 log, requiring improvements in cell culture and purification methods. We needed a formulation that would not require drug-product (DP) storage in a deep freeze. Finally, GSK would have to develop and qualify in-process analytical characterization methods for quality control (QC), with improved robustness that could support the release of cell banks, virus seeds, drug substance (DS), and DP in accordance with regulatory agency guidelines.
The primary challenges for the DP team were to stabilize ChAd3-EBO-Z at 2–8 °C and to rapidly develop process analytical tools and knowledge. Frozen storage is very difficult for shipping products to and throughout Africa. A buffer previously developed by Merck that could stabilize human adenovirus in a liquid state for several years was inappropriate for chimpanzee adenoviruses. However, because of its ethanol and high salt content, it is not the best choice for freezing or trying to develop a freeze-dried formulation.

Table 1: Main process analytical technologies (PATs) used during drug product development; GFP = green fluorescent protein
Analytical Development
Before working on thermostabilization of the vector, we focused on quality and analysis. We identified three critical quality attributes (CQAs) for this vaccine product: physicochemical integrity of the virus, functional infectivity (live virus), and particle content (viral quantity). Table 1 summarizes the tools we developed to give us results within a one-week timeframe for monitoring those parameters and to allow sustainable iterations throughout the DP development.
For physicochemical integrity, we monitored the size and degradation of virus particles using dynamic light scattering (DLS). We measured capsule integrity with a PicoGreen assay (Thermo Fisher Scientific), which is based on fluorescent staining of double-stranded DNA. When virus capsules are disrupted, they release DNA that fluoresces.
To monitor functional infectivity, we use two techniques. Cell culture infectious dose (CCID50) testing would not work for large numbers of samples, so we developed a higher throughput test based on fluorescence-activated cell sorting (FACS) that provides results in one day.
Finally, for particle counting, we used quantitative polymerase chain reaction (qPCR) to measure the number of copies of a cytomegalovirus promoter inserted into the viral genome. Later, we shifted to analytical ion-exchange chromatography for this parameter.
Formulation Development
Our purpose was to develop a lyophilized product, so we first looked at the glass-transition temperature of the formulation. With the original buffer, Tg′ was –51 °C, which is completely incompatible with lyophilization. To make it more suitable, we removed ethanol and ethylenediaminetetraacetic acid (EDTA) from the buffer ingredients and decreased the salt concentration while increasing sucrose to keep the formulation isotonic. The modifications improved Tg′ to –35 °C, which was now compatible with lyophilization cycles.
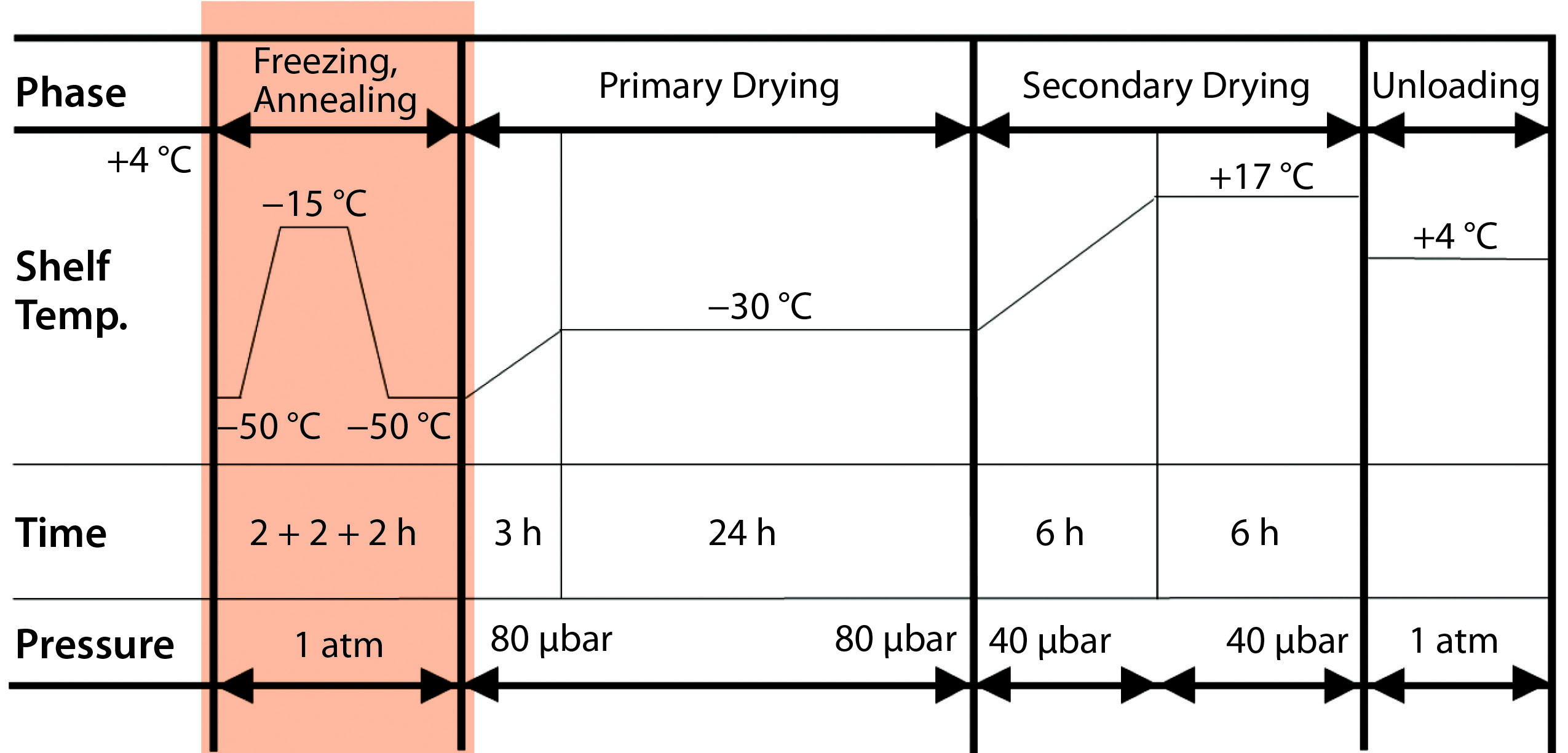
Figure 1: Impact of annealing on the freeze-drying cycle. Mathot F, Bourles E. Pharmaceutical Composition Comprising an Adenoviral Vector. WO 2017013169 A1 26 January 2017.
With the new “light” buffer, we investigated two lyophilization process variants, with and without an annealing step. That is when, during the freezing phase of freeze-drying, a product is maintained at a specified subfreezing temperature (slightly above Tg′) for a predetermined time period. That can cause Ostwald ripening of ice crystals and cryoconcentration of an amorphous matrix. Figure 1 shows the lyophilization cycle we applied, with annealing at the end of the freezing step, followed by primary and secondary drying.
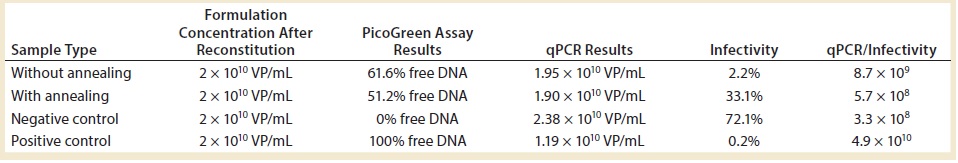
Table 2: Impact of annealing step on the freeze-drying cycle; negative control is the starting adenoviral stock; positive control is the same material intentionally degraded for 30 minutes at 60 °C.
Table 2 shows the results. For every condition, we tested two controls: a freshly thawed DS diluted at the DP concentration to get a negative control and the same submitted to full degradation of 30 minutes at 60 °C for a forced-degradation positive control. We were surprised to find that the best conditions came with inclusion of the annealing process. PicoGreen assay results fell in the midrange (the lower, the better), and infectivity was ~30% (the higher, the better). Without annealing, the latter was only 2%. We hypothesize that this virus is very sensitive to cold denaturation (which makes sense for a virus that evolved to infect chimpanzees in Africa), and the annealing step decreases the interfacial surfaces that come into contact with it.
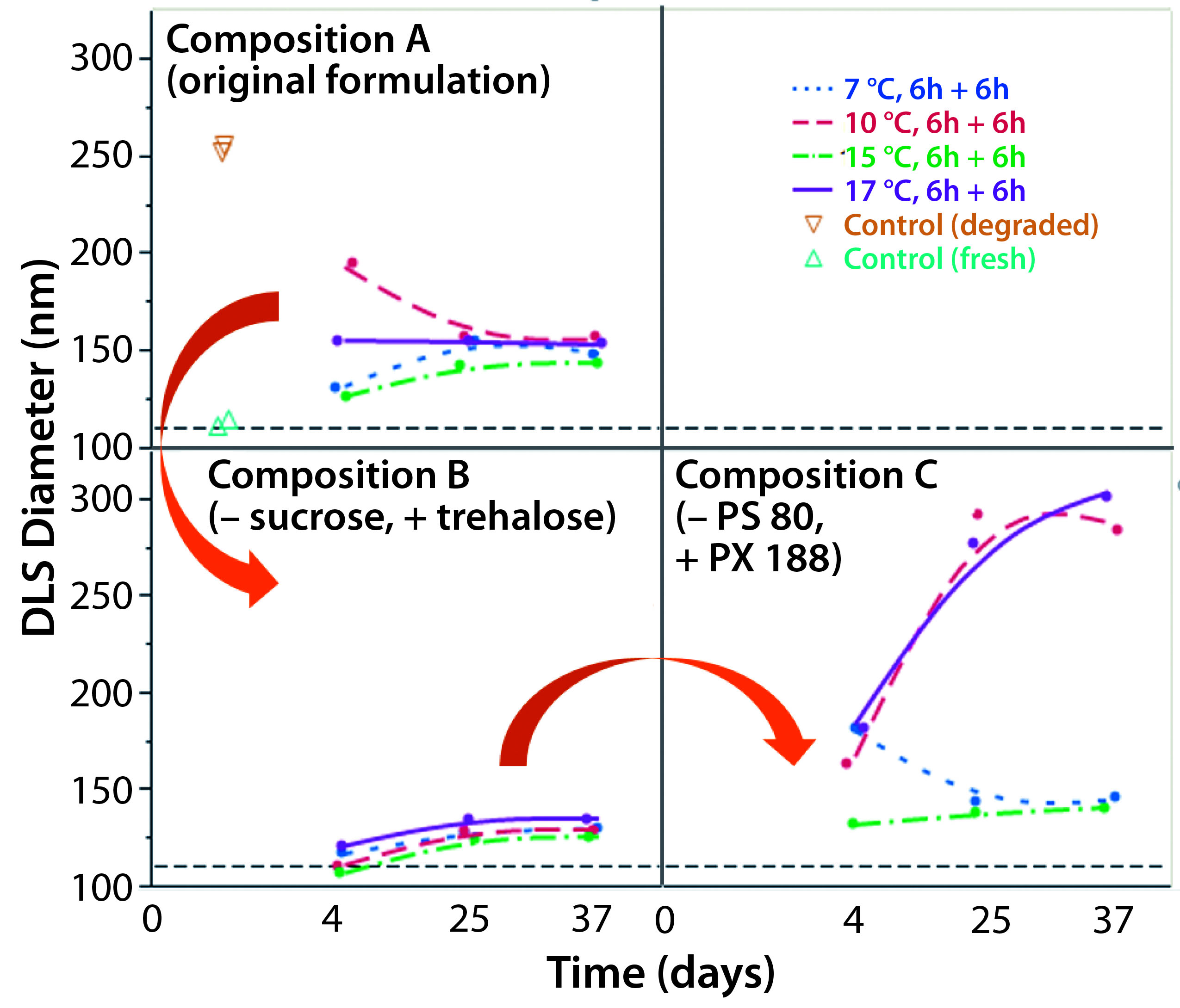
Figure 2: Replacing sucrose with trehalose and adding PS 80 as a surfactant provided the best stabilization. Composition A: ChAd3-EBO-Z 1.1011 VP/mL; Tris 10 mM, pH 7.5; histidine 10 mM; NaCl 25 mM; sucrose 8%; MgCl2 1 mM; polysorbate 80, 0.02% Composition B: ChAd3-EBO-Z 1.1011 VP/mL; Tris 10 mM, pH 7.5; histidine 10 mM; NaCl 6 mM (residual); trehalose 7%; sucrose 2% (residual); MgCl2 1 mM; polysorbate 80, 0.02% Composition C: ChAd3-EBO-Z 1.1011 VP/mL; Tris 10 mM, pH 7.5; histidine 10 mM; NaCl 6 mM (residual); trehalose 7%; sucrose 2% (residual); MgCl2 1 mM; poloxamer 188, 0.15%
Next, we tried to improve the lyophilization matrix by revisiting the lyoprotectant and surfactant composition in the buffer. We replaced the sucrose with trehalose and polysorbate 80 with poloxamer 188. When looking at the virus size by DLS as a CQA (Figure 2), we found the best stability came when sucrose was replaced by trehalose, with polysorbate 80 as a surfactant. Indeed, massive aggregation was seen when we used the poloxamer 188 during the lyo step. So we decided to keep the polysorbate 80 as a surfactant and further investigate whether trehalose was the best choice with it.
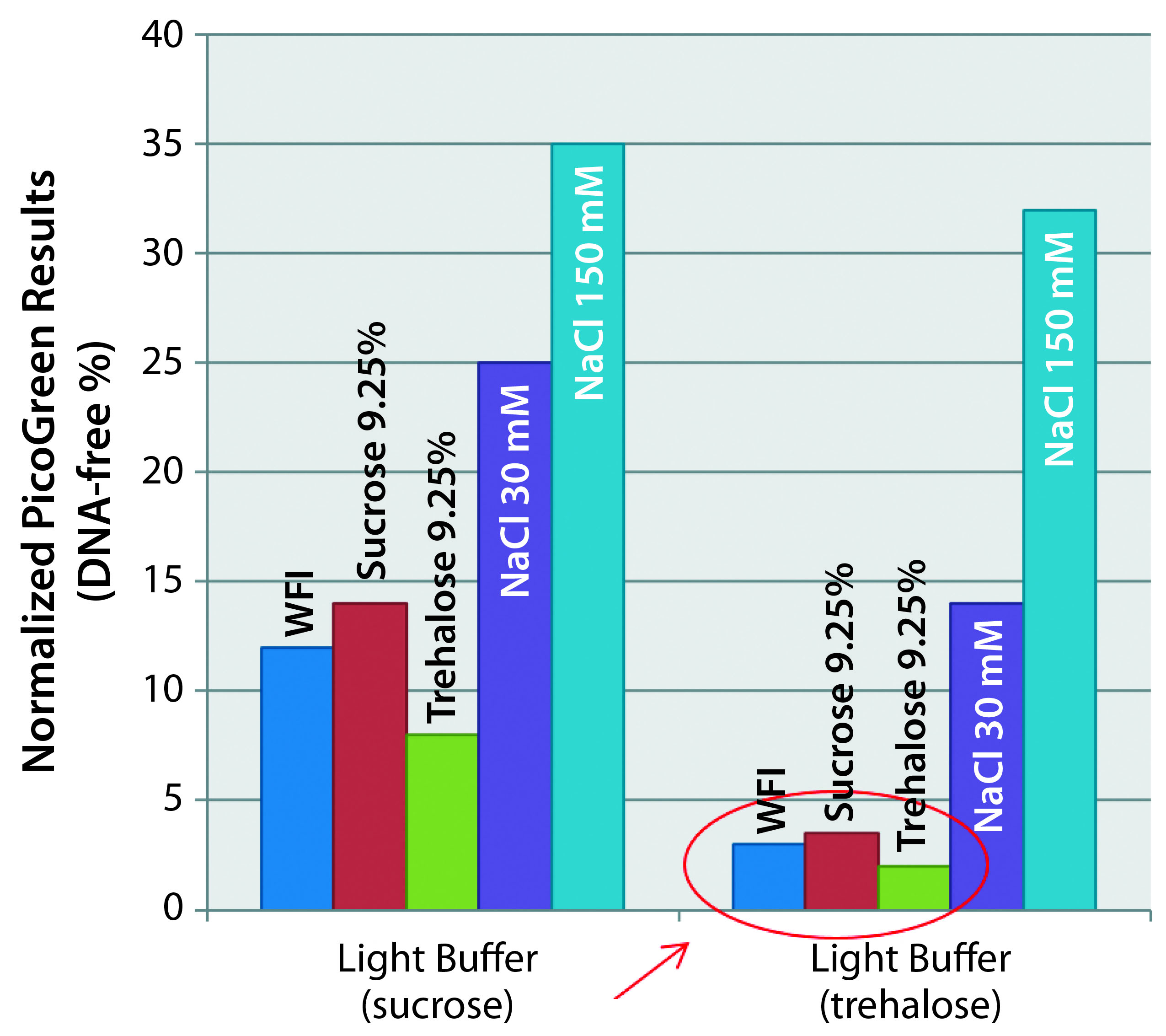
Figure 3: Matrix optimization — impact of reconstitution medium’s salinity (different rehydration media tested to reconstitute freeze-dried vaccines incubated for two months at +4 °C). Preserved capsid integrity when NaCl-free media are used with the trehalosebased lyophilization matrix. Hypertonicity has no effect as long as NaCl-free media are used.
For that purpose, we produced two variants of the light buffer: one with sucrose, the other with trehalose. And we incubated drug products with each for two months at 4 °C, then hydrated them with different media. We tried water for injection (WFI), isotonic sucrose, isotonic trehalose, and two concentrations of NaCl (30 mM and 150 mM). Figure 3 shows the PicoGreen assay results that followed. (Again, the lower numbers are better here, showing no capsule disruption.) The best results came when we used a salt-free medium and with the trehalose base matrix. Based on those results, hypertonicity has no impact as long as a salt-free medium is used. We do not have an explanation for that yet, but we do know that sugar has unique glass-transition properties (much greater than other regularly used disaccharides).
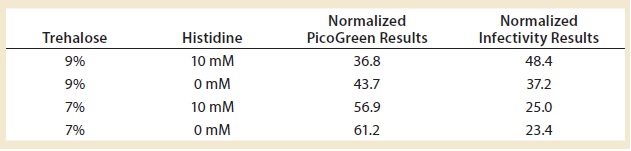
Table 3: Fine-tuning the lyophilization matrix — investigating trehalose concentration and the effect of histidine
Finally, we tried to further optimize the lyophilization matrix by investigating higher concentrations of trehalose with and without histidine, which is a known stabilizer for adenoviruses (4). We found that the optimal conditions — determined by PicoGreen and infectivity assays — included both trehalose and histidine (Table 3). Infectivity was preserved at ~50% (a 0.3-log loss), meaning that half the product was completely destroyed after freeze-drying. In the field of live-virus vaccines, a 0.3-log loss is acceptable. But because we were responding to a health emergency, we could not sacrifice half of our production: Every dose produced was needed.
A Work in Progress
We have successfully improved the development of this lyophilized ChAd3-EBO-Z vaccine for the African market. From a 1-log product loss at the start, we achieved 0.3 log loss. We found it interesting that this adenovirus is very sensitive to the presence of NaCl and also that, by including trehalose in conjunction with polysorbate 80 surfactant, we could drastically improve product stability. An annealing step during lyophilization brings superior performance to virus stability and infectivity for this vaccine.
However, this version of the vaccine is not fully acceptable for a health emergency because of the induced process loss. We found a compromise for production of Ebola health emergency lots using a back-up solution (investigated in parallel) that requires keeping the vaccine at ≤–60 °C but allows for temperature excursions up to –20 °C for up to 10 months (thanks to the light buffer formulation) with no loss in infectivity. That makes it easier to ship to and around Africa for storage in dispensaries.
The story is not over for our lyophilized formulation of adenoviral vectors, however. We continue to develop freeze-dried vaccines as well as liquid dosage forms.
| Questions and Answers |
| This article is adapted from a presentation by Mathot at the BioProcess International Conference and Exhibition in Boston, MA, on 28 September 2017. Several audience members posed questions and offered comments after the talk. |
| Did you find correlation between moisture content and stability? Yes. It’s not presented here, but we have proved during further development that residual moisture is a key element of stabilizing this virus. So if we want to keep a certain infectivity, we can’t dry the formulation too much. |
| Did you use the same container– closure for 2–8 °C, –20 °C, and –80 °C storage? Yes, we used the same stoppers, capsules, and vials, and we have specific testing for the container–closure integrity. |
| Did you try a larger molecule than trehalose/sucrose for cryo-preservation? We are limited to certain excipients for injection to humans. Very large molecules are not allowed because of renal toxicity. You can drastically increase the Tg’ of a product by using a high–molecular-weight polymer, for instance. But we are restricted to a certain panel of excipients for this type of product, so for the moment we are not investigating that.
In principle, you could use them and then simply digest them to remove or convert them into monomers. That could be a solution, yes. |
| Why didn’t you completely get rid of the salt in the first screening, when you went to sucrose and NaCl at 25 mM? You might have brought Tg’ up a bit more. I didn’t mention that we did another screening in which we saw that we had to maintain a certain amount of NaCl for stability of this charged virus. |
Acknowledgments
We acknowledge the work of GSK’s lyophilization and formulation teams in Belgium: Delphine Guillaume, Catherine Van Loo, Marie-Hélène Delannoy, Stéphane Godart, Vincent Ronsse, Olivier Despas, Alain Philippart. In collaboration with the Vaccine Research Center (VRC) for the development of the Ebola candidate, this work was sponsored and financially supported by GlaxoSmithKline Biologicals SA, the US Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA), and the Bill and Melinda Gates Foundation.
References
1 Stanley DA, et al. Chimpanzee Adenovirus Vaccine Generates Acute and Durable Protective Immunity Against Ebolavirus Challenge. Nat. Med. 20(10) 2014: 1126–1129; doi:10.1038/nm.3702.
2 Cottingham MG, et al. Preventing Spontaneous Genetic Rearrangements in the Transgene Cassettes of Adenovirus Vectors. Biotechnol. Bioeng. 109(3) 2012: 719–728; doi:10.1002/bit.24342.
3 Rappuoli R, De Gregorio E. Vaccines: Novel Technologies for Vaccine Development. Curr. Opin. Immunol. August 2016; v–vii; doi:10.1016/j.coi.2016.07.001.
4 Evans RK, et al. Development of Stable Liquid Formulations for Adenovirus-Based Vaccines. J. Pharm. Sci. 93(10) 2004: 2458–2475; doi:10.1002/jps.20157.
Frédéric Mathot, PhD, is senior scientist; and Erwan Bourlès, PhD, is an expert scientist in drug-product technical research and development at GSK in Rixensart, Belgium. The authors are listed as inventors on an international patent application in the name of GlaxoSmithKline Biologicals SA: WO2017013169 A1, “Pharmaceutical Composition Comprising an Adenoviral Vector,” 20 July 2016. GlaxoSmithKline Biologicals SA was involved in all stages study conduct and analysis.