Over the past few years, Oxford Biomedica Ltd. (OXB) has developed and implemented a fill–finish platform (“Oxbox,” Figure 1) at its viral vector processing facility in the United Kingdom. The facility includes four segregated bulk viral-vector drug substance (VS) suites, where closed systems and bioburden control processes apply, and two viral-vector drug product (VP) fill–finish suites that apply aseptic processing, with space for expansion by scale-out as product output demand increases. Segregated suites enable the facility to process different viral vectors simultaneously, including OXB’s own LentiVector delivery platform and other types of viral vectors, by acting as a contract development and manufacturing organization (CDMO) through strategic partnerships.
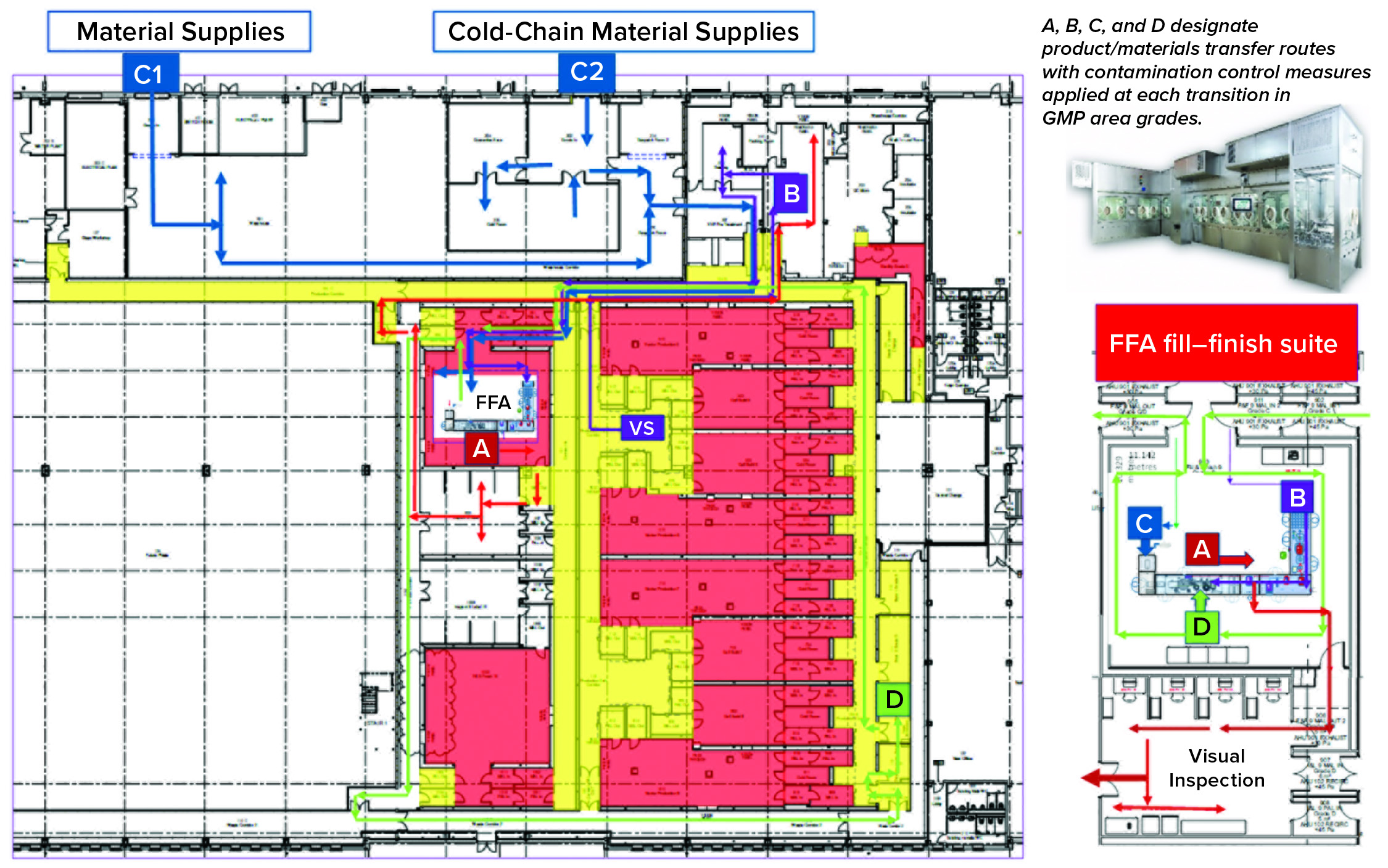
Figure 1: Oxbox viral vector facility at Oxford Biomedica UK
The Oxbox processing platform combines a VS pooling stage that applies bioburden control with a VP concentration process that incorporates single-use closed systems followed by automated vial filling and container–closure within barrier isolator technology under full aseptic processing conditions. The platform is flexible enough to be configured for filling viral vectors under different conditions, covering a wide range of volumetric concentration factors and vial-size–fill-volume combinations. VS and VP processed within the Oxbox platform are used either for gene transfer to support gene-modified autologous or allogeneic cell therapy — or for direct application as an in vivo gene therapy drug product.
Part 1 of this two-part case study focuses on the design of the formulation–fill–finish process and facility itself. In BPI’s November–December 2021 issue, we will conclude with a discussion of problems solved, decisions made, risk management, and viral safety concerns.
| Overview of Oxford Biomedica |
| Advanced-therapy medicinal products (ATMPs), specifically viral-vectored gene and gene-modified cell therapies, are the business focus of Oxford Biomedica (OXB). Using its unique LentiVector delivery platform, OXB has created an extensive portfolio of product candidates for treating oncological, ophthalmological, and central nervous system (CNS) disorders.
OXB also has strong partnerships in place with a number of product developers, including large pharmaceutical companies such as Bristol-Myers Squibb (BMS) and Novartis, for which it serves as a contract development and manufacturing organization (CDMO). OXB is the global supplier of the vector used in manufacturing Novartis’s Kymriah (tisagenlecleucel), the world’s first commercially approved chimeric-antigen receptor (CAR) T cell therapeutic product, which is approved in multiple territories across the globe. OXB also partners with early stage biotechnology companies developing novel gene and cell therapies. The company continues to invest in innovation — including platform technologies and product research and development — with a view to developing new ATMP products targeting unmet clinical needs. |
Viral Vector Product Requirements
Development of this flexible combined formulation and filling platform was driven by the need to meet full good manufacturing practice (GMP) compliance. The EU GMP Annex 1 for manufacture of sterile medicinal products, EU GMP Part 4 for advanced-therapy medicinal products (ATMPs), and US Food and Drug Administration (FDA) guidance for sterile products made by aseptic processing provided key information. To meet those needs, the company applied advanced technology and alternative processing methodologies that follow quality risk management (QRM) principles together with viral containment (to prevent environmental release) as part of a contamination control strategy (CCS) for processing a genetically modified organism (GMO) with cross-contamination control.
Demand is increasing for gene and cell therapy processes with higher yields, as evidenced by a number of recent high-profile approvals and an increasing number of products in clinical development for new target patient populations. In response, the industry needs to develop new processing methodologies that maintain product quality, particularly in regard to sterility assurance.
Closed-system processing often is used during production of cell-based ATMPs: cell culturing, purification, and subsequent manual process manipulations occur within microbiological safety cabinets (MSCs). Such an approach often is used for preparation of autologous (single-patient) therapies and for some viral-vector–based cell processing for early phase clinical development.
When open processing steps are used in MSCs, products or substances are exposed for limited periods to the processing environment. Thus, the approach provides only limited protection of products from extraneous contamination, which (if present) could harm patients and/or compromise product efficacy. MSCs are designed for operator safety through containment of air flow as well as maintenance of a clean-air environment for applications such as cell/tissue culture. OXB has adopted closed-system processing wherever possible to limit potential environmental exposure of the low-bioburden VS to a small number of open processing steps.
In designing the Oxbox facility, the company understood that such an approach is suitable for VS production. However, using knowledge gained in developing previous VP processes with contract manufacturing partners, OXB chose to maintain appropriate sterility assurance with a more sophisticated approach to VP processing after sterilizing filtration. Regulators encourage development of appropriate approaches for GMP-compliant batch manufacturing for aseptic processing stages — e.g., filling or other aseptic processing steps performed after sterilizing filtration — that adopt advanced barrier–isolator technology with integrated automation. Such approaches support regulatory expectations for separation of people and processes, with process design following QRM and quality by design (QbD) principles to apply the technology as a technical contamination-control measure. In addition, regulators require GMP manufacturers to follow scientific and technical progress, generally encouraging adoption of new technologies (1).
Continued development of gene and cell therapies to target unmet patient needs — in particular for indications with increased demand — requires new approaches that provide improved risk management with assurance of sterility. When a viral vector will be used in further processing for an ex vivo gene therapy — e.g., based on chimeric-antigen receptor (CAR) T cells — it will not be administered to patients directly or immediately. Such cases, however, bear the same expectation of sterility assurance that is required for directly administered products (e.g., viral vectored gene therapies).
Therefore, our approach has been to develop an aseptic processing and filling capability targeting the highest standards of sterility assurance to serve both.
For maximum assurance of sterility in fill–finish operations, sterile processing intermediates are exposed to a grade A processing environment. Current thinking is to move away from highly manual MSC-based filling processes toward adopting automated aseptic-processing equipment and barrier technologies: isolators and restricted-access barrier systems (RABS). Viral vectors pose an increased difficulty in processing: Such products often are frozen or cryopreserved, with a necessary “thaw-to-freeze” stability processing time window for completion of all processing steps from bulk formulation through filling, visual inspection, labeling, and secondary packaging. Filling is just one part of that process, which must be considered holistically so that the entire process will be efficient, and each step can be integrated with the next as quickly and simply as possible.
Cleaning Residuals: Another principal challenge in aseptic processing comes with residuals from disinfection agents (including sporicides and biocides) used in biodecontamination of processing environments and integrated equipment. For example, residual hydrogen-peroxide vapor (VHP or vH2O2) can compromise the function (efficacy) of biological products. Enveloped viral products (e.g., lentiviral vectors) in particular are highly sensitive to the oxidizing potential and free-radical attack mechanism of H2O2.
VHP also is applied as an effective cross-contamination control measure for viral clearance between different products. The general consensus for conventional/chemical medicinal products is to purge H2O2 to a background level of 1 ppm residual before processing begins. However, for viral vectors, a significantly lower background level (0.1 ppm) is preferred to mitigate the risk of product inactivation.
The Oxbox case study was originally presented at the International Society of Pharmaceutical Engineers (ISPE) 2020 Aseptic Conference (2), with a follow-up given in 2021 (3) after the filling platform was installed, with qualification progressing toward aseptic process simulation with media fills.
Facility and Process Design: Key Drivers and Decisions
The Oxbox facility design was adapted from a large existing building that originally had been constructed and used as a postal sorting office. The 7,800-m2 open-plan facility has very few supporting pillars and a significant height (>9 m) to the eaves. Such a space provided the perfect shell for a purpose-built processing facility for VS/VP manufacturing. This approach reduced overall project execution time, allowing partial occupation of the site at an early stage of the project to facilitate direct on-site interaction with architects, engineers, and construction teams.
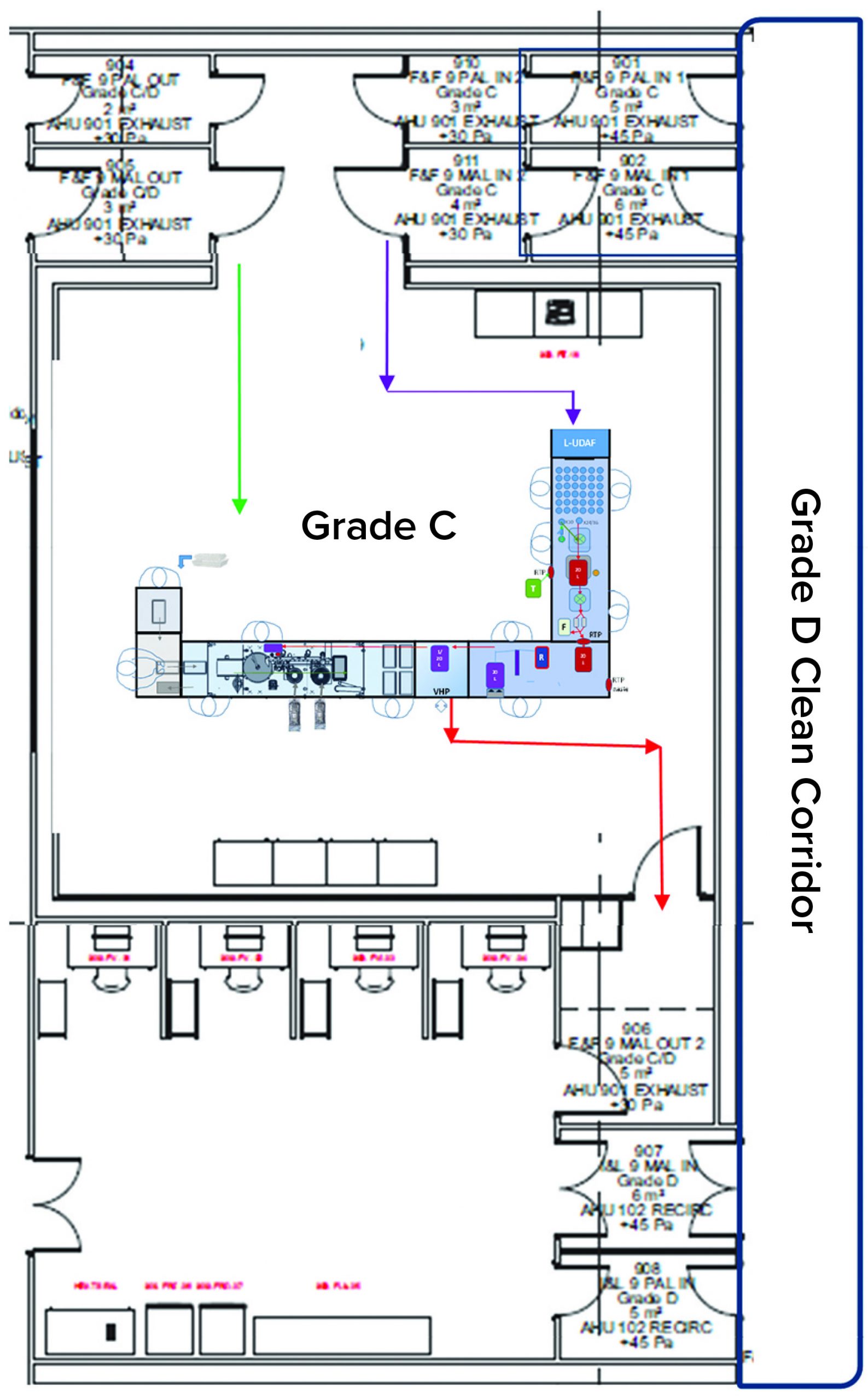
Figure 2: Fill–finish suite
Key Decisions in Process Design: From the outset, the facility design was based on dedicated suites with independent air-handling units to provide area-process segregation, each with airlocks to enable a pressure cascade incorporating negative sinks and positive bubbles for viral containment and cross-contamination control. Corridors connected different suites and centralized areas: warehouse, microbiology laboratory, general change area (from street access), and so on. Design of the suites was based on a fundamental philosophy for unidirectional flows of people, materials, products, and waste. That was achieved by combining separate entry and exit material and personnel airlocks (MALs, PALs) and pass-through hatches for samples and process intermediates. Products and waste exiting the cleanrooms through MALs were segregated through separation in time.
Four independent VS suites were constructed for processing of fresh or frozen viral vector substances. Where required, vector substance thawing is performed in a controlled process area outside the VS suites, with thawing of closed containers completed in a water-for-injection (WFI) bath.
The facility was designed with two fill–finish suites. Each of those comprises a GMP grade C filling room with barrier technology and a separate area housing visual inspection booths (4, 5) in a clean, nonclassified (CNC) area that has a dedicated grade D MAL from the filling room to receive/transfer filled products. Labeling and secondary packaging equipment also was installed within the visual-inspection room so that specific products could be processed within each suite with cross-contamination control measures provided between different filling suites.
Two side-by-side fill–finish suites initially were built on the same design, thereby providing the flexibility to install different filling technology platforms as required in the future. Key to the facility and process design was supporting the concept of scale-out rather than scale-up to increase product output capacity. Space for expansion is provided to duplicate identical qualified processing suites, including qualified technology with corridors that link to the centralized facilities.
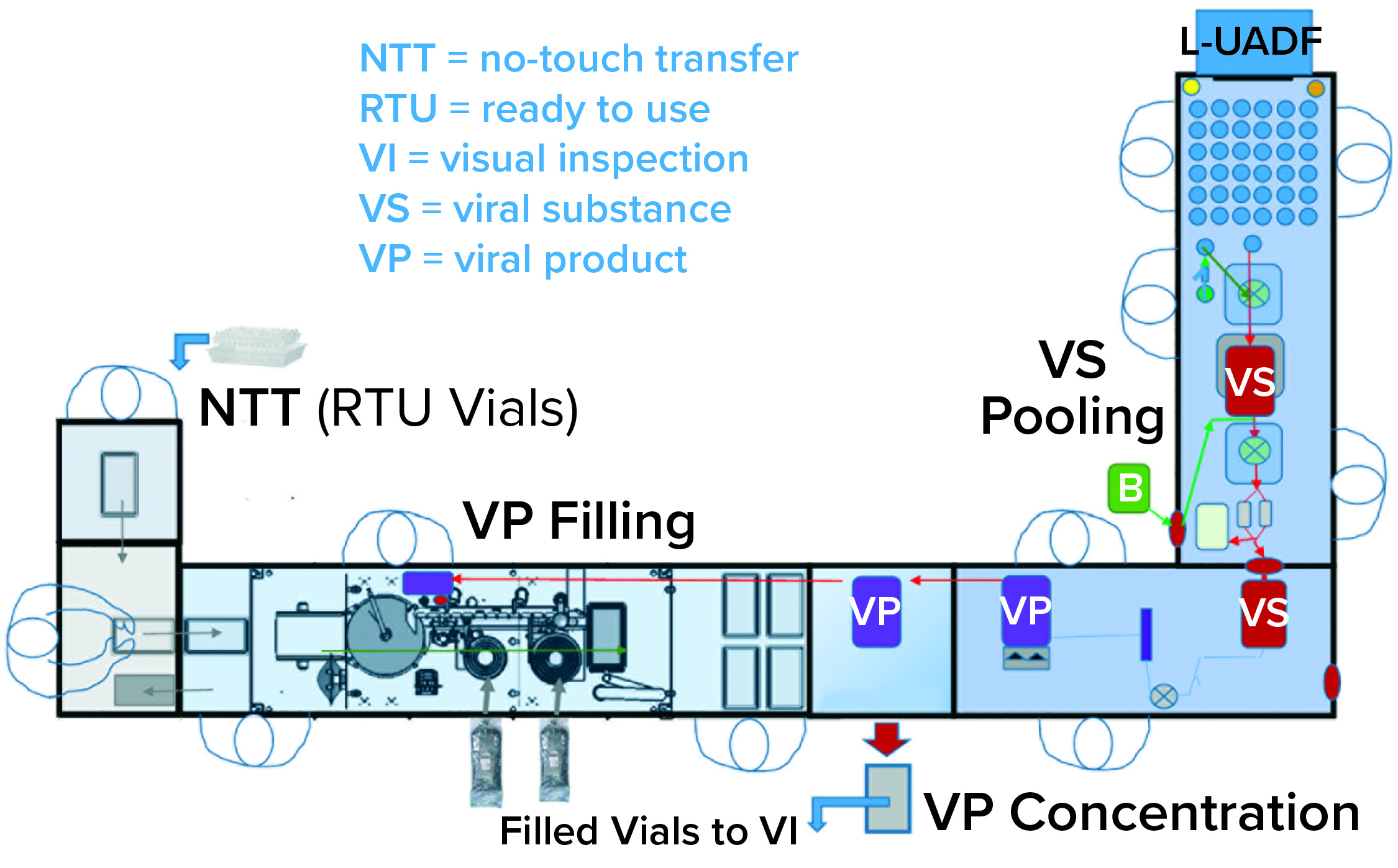
Figure 3: Combined viral vector formulation and filling platform
One of the two filling suites is complete and fully equipped, with the second line planned to be equipped based on demand. Requirements for the fill–finish platform were sent to different suppliers, and their responses were reviewed thoroughly. OXB selected barrier technology from Franz Ziel GmbH and filling technology from Watson-Marlow Flexicon A/S. Their designs offered the greatest flexibility to enable adaptability and continued development of the filling processes. Figures 2 and 3 show the layout of the combined formulation and filling line, which provides significant process benefits related to speed and safety. Interconnected process zones are based on isolator barrier technology with closed material transfer systems. Those provide for viral containment within isolators and facilitate rapid processing/transition between process stages to satisfy process time limitations.
Finding Flexibility: Single-use systems (SUS) and ready-to-use (RTU) presterilized product containers (vials) and closures provide necessary flexibility. Based on barrier technology within a dedicated fill–finish suite, the combined formulation and filling platforms (Figure 3) were specified to fill RTU product containers (class 1 borosilicate vials) and closures (elastomeric stoppers and aluminum crimps with flip-off caps). Product and buffer bulk containers (bags) and fluid paths, including filling needles, are presterilized SUS.
The OXB VS process has been optimized to reduce the volumes of buffers and WFI required (production contracted out). The Oxbox facility was designed with space for an on-site WFI generation system; however, installation of the system was deferred to future phases until demand for volume increases. Because the facility currently does not have a WFI system installed to supply an autoclave, the decision was to focus on VS/VP manufacturing activities using presterilized units so that sterilization processes also would be contracted out.
The one exception to that policy is related to sterilization of parts that come into direct contact with product. Those include parts that contact the stoppers (bowl, chute, rail, and holding jaws) for which the regulatory expectation is sterilization out-of-place. Bioburden control is achieved through transfer and assembly followed by VHP in place within the isolator barrier technology before aseptic process filling begins (4, 5).
The direct–product-contact parts required reprocessing (cleaning sterilization) before use in batch manufacturing operations. Thus, on-site sterilization was required. Those parts are relatively small and machined out of solid pieces of 316L stainless steel that won’t melt or warp when exposed to high temperatures. So they are suitable for dry-heat sterilization, which OXB decided would be easier to manage on site than it would be to specify a moist-heat sterilization autoclave. The dry-heat oven can operate at the higher temperatures for depyrogenation, so the process also is qualified for depyrogenation to provide an endotoxin control measure for the direct-contact parts.
The dry-heat oven is situated in an ancillary grade D cleanroom away from the grade C filling suite. Key to reprocessing of the direct-contact parts with end-to-end assurance of sterility during transfer operations from the sterilization/depyrogenation area to the filling suite was sourcing and application of heat-resistant packaging. That can be closed (by heat sealing) to the external environment to maintain a sterile barrier until the parts are placed into a isolator. By contrast, the capper bowl is not considered to be a direct-contact part, so its assembly was subjected only to VHP cleaning in place.

Direct product-contact parts in the filling isolator
No-Touch Transfer: Because residual VHP can affect viral vector function, an alternative methodology was required for transfer of presterilized RTU containers into the grade A filling zone. No-touch transfer (NTT) was specified because it requires no disinfection agent applied during transfer into the grade A isolator. NTT has been developed and qualified as an alternative methodology with semi- and fully automated debagging technology that follows both QRM and GMP principles without a disinfection step applied during its two debagging transfer steps (6).
RTU containers (vials) within a sterile barrier (a plastic tray sealed with a Tyvek cover) are qualified in manufacturing so that the outside of the sterile barrier is also sterile (suitable for the NTT process) and remains so through the supply chain until point-of-use NTT transfers. Through in-process NTT transfers on the filling line, the sterile barrier never gets exposed to an area below grade A. That provides a rationale to justify the elimination of a disinfection step for the outer packaging during material transfer from the grade C processing room into the grade A isolator through the NTT.
That approach offers an added benefit of shortened production start-up times. With no VHP used in material disinfection of the sterile barrier for the RTU vials (which are permeable to VHP), aeration times did not have to account for vial degassing. Note also that NTT enables on-demand feed of sterile containers to support increased fill-line speeds and batch sizes with no requirement to preload the isolator with all vials before processing can start.
Looking Ahead
Part 2 will conclude this report in the next regular issue of BioProcess International. It includes discussion of problems solved, decisions made, risk management, and viral safety concerns.
References
1 Article 23. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use. Off. J. Eur. Union 311, 28 November 2004: 67–128; https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/directive-2001/83/ec-european-parliament-council-6-november-2001-community-code-relating-medicinal-products-human-use_en.pdf.
2 Bull M, Drinkwater J. Oxford Biomedica UK Integrated Aseptic Processing Platform for Multiple ATMP Indications Including a Case Study on a Viral Vector Used in a CAR-T Cell Cancer Therapy. ISPE Aseptic Conference, 18–19 March 2019, Rockville, MD. International Society for Pharmaceutical Engineering: North Bethesda, MD; https://ispe.org/conferences/2019-aseptic.
3 Southam L, Drinkwater JL. Combined Formulation–Fill–Finish of ATMPs: Lentiviral Vector: Oxbox Case Study. ISPE Aseptic Conference, 2–3 March 2020, North Bethesda, MD. International Society for Pharmaceutical Engineering: North Bethesda, MD; https://ispe.org/conferences/2020-aseptic.
4 Hopkins A. VHP (Vapour Hydrogen Peroxide) Fragility. MHRA Inspectorate Blog 20 April 2018; https://mhrainspectorate.blog.gov.uk/2018/04/20/vhp-vapour-hydrogen-peroxide-fragility.
5 Clarity on GMP Guidance Note No. 1: Assurance of Sterility for Container Closure In-Direct Product Contact Surfaces in Aseptic Process Filling. Pharmaceutical and Healthcare Sciences Society: Swindon, UK, 2018; https://cdn.ymaws.com/phss.co.uk/resource/resmgr/files/phss_clarity_on_gmp_guidance.pdf.
6 Drinkwater JL, et al. No-Touch-Transfer (NTT) Pre-Sterilised Container Entry into EU Grade A Filling Environments Following GMP and QRM Principles. Eur. J. Paren. Pharm. Sci. 23(2) 2018: 43–52; https://www.researchgate.net/publication/327939939_No-touch-transfer_Pre-sterilised_container_entry_into_EU_grade_a_filling_environments_following_GMP_and_QRM_principles.
Leslie Southam is the quality assurance manager of the Oxbox project at Oxford Biomedica Ltd. in Oxford, United Kingdom; 44-1865-783-000; https://www.oxb.com. Based in the United Kingdom, corresponding author James L. Drinkwater is head of GMP compliance for Franz Ziel GmbH and elected chair of the Pharmaceutical and Healthcare Sciences Society (PHSS); james.drinkwater@ziel-gmbh.com; https://www.ziel-gmbh.com.
LentiVector is a registered trademark of Oxford Biomedica Ltd. Tyvek is a registered trademark of DuPont de Nemours, Inc. Kymriah is a registered trademark of Novartis AG.