Cell-based potency testing provides quantitative data concerning a drug’s biological activity. Thus, it plays an essential role in biopharmaceutical quality control (QC), good manufacturing practice (GMP) product release, comparability determination, and stability testing for both drug products and drug substances. Potency is a critical quality attribute (CQA) often scrutinized by regulators and reviewers. Test methods are specific to a drug’s mechanism of action (MoA) and should be validated to internationally harmonized regulatory standards (1). The options preclude applying a simple one-size-fits-all template for automation of potency assays — typically enzyme-linked immunosorbent assays (ELISAs) — but the concept is well worth consideration.

Figure 1: Efficiency gains provide time savings, but not in a simple way for potency assays; e.g., incubation times don’t change; HoT = hands-on time.
At the CASSS Well-Characterized Biotechnology Products (WCBP) conference in May 2019, Hermann Beck (F. Hoffmann-La Roche) presented an approach to automation of potency assays under the rubric of “It’s not plug-and-play; it’s a journey.” In 2020, we discussed the topic further.
When it comes to automation, there are three kinds of bioassayists: those who use it, those who don’t, and those who wonder which way to go. “Shall we use it or not?” they ask. “Will it pay off for us? And what can we expect if we decide to automate?”
Automation is commonly used in biopharmaceutical laboratories, Beck explained, for high-throughput tasks such as screenings and diagnostics. But when does it make sense to automate bioassays for QC? He listed a few typical arguments for doing so, from simple ergonomics and efficiencies to lowering failure rates and facilitating method transfer. But the question isn’t that simple and should be considered strategically.
Balancing Benefits and Effort
Microplate-based bioassays involve a great deal of pipetting — as many as 400 such fluid transfers in an average potency assay. Nearly half of technicians surveyed in two QC bioassay laboratories at Roche reported repetitive-motion issues related to such work, providing a strong motivation for letting machines do it. Beck also referred to “heterogeneous work packages,” in which several different steps need to be performed for a given assay. “You do microscopy and handle culture flasks, run a centrifuge, and use all kinds of standalone devices. Some of these steps can be designed for automation easily, some are difficult but can be automated, and some steps are very difficult or cannot be automated with reasonable effort.”
Efficiency Gains and Time Savings: Figure 1 compares typical manual and automated bioassay workflows. Both include some hands-on time (HoT, in gray), and the manual workflow includes some time when laboratory capacity is free (blue) as well as some tests that could be automated (green). Because of incubation requirements, the overall assay duration is the same. So you save no time there, but HoT can be shortened. Calculating that difference is a simple way of making a business case. If you multiply that HoT reduction over many assays per year, the time savings can seem to be substantial. “But I think such an approach is too simple,” said Beck.
Setting up and maintaining laboratory automation takes effort. QC groups must balance the ergonomic benefits and reduction of HoT and complexity against installation and maintenance, equipment qualification, method programming, and validation. Reduction of errors is a benefit, as well. To address the question of whether automation will pay off for a given company, a number of factors become relevant: the number of samples run per time period, the number of different assays used, the extent to which those tests can be automated, and the number of laboratories involved.
Throughput, Beck said, is a major issue. “Does automation make sense only for high-throughput tasks?” He explains that decision-makers need to consider the number of samples and the number of different assays as well as associated time constraints. At one extreme, a laboratory might perform just few different assays on a large number of samples; at the other would be many different assays using few samples. “You have high throughput on one hand,” as Beck characterized it, “and high diversity on the other.”
A case could be made for automating either one — although it’s an easier argument for high-throughput assays. “But it would make sense also for rare assays,” Beck explained. It’s hard to develop a routine for those applied only once in a while (e.g., twice a year). “That means you always need a refamiliarization, which in turn increases failure rates and requires more time and training. Here, automation could be a tool to cope with high diversity as well as high throughput.”
Transferability: Another important consideration is the number of different laboratories that will be performing a given assay. For any number more than one, method transferability becomes important. Large organizations can face great complexities, with several different laboratories running different automation systems from different suppliers, or the same equipment set up in different configurations. If those teams want to exchange an automated method, that process is unlikely to run smoothly. Reprogramming and reconfiguration could be necessary: A team borrowing an automation method would need to start over from scratch during its application because direct exchange is impossible.
To prevent such situations, Roche implemented a company-wide approach within bioassay testing a few years ago. “We did not want to be reinventing the wheel all the time, with every lab duplicating effort and ending up with isolated solutions. This was the starting point then for our global bioassay automation team.” The company has bioassay laboratories in Germany, Switzerland, and California. “The focus of this team was to exchange information and harmonize a potency assay automation solution.” The goals were to facilitate transferability and provide consistent approaches to qualification and other GMP issues, such as reporting. “These are all things that, once developed, everyone can use without having to find individual solutions.”
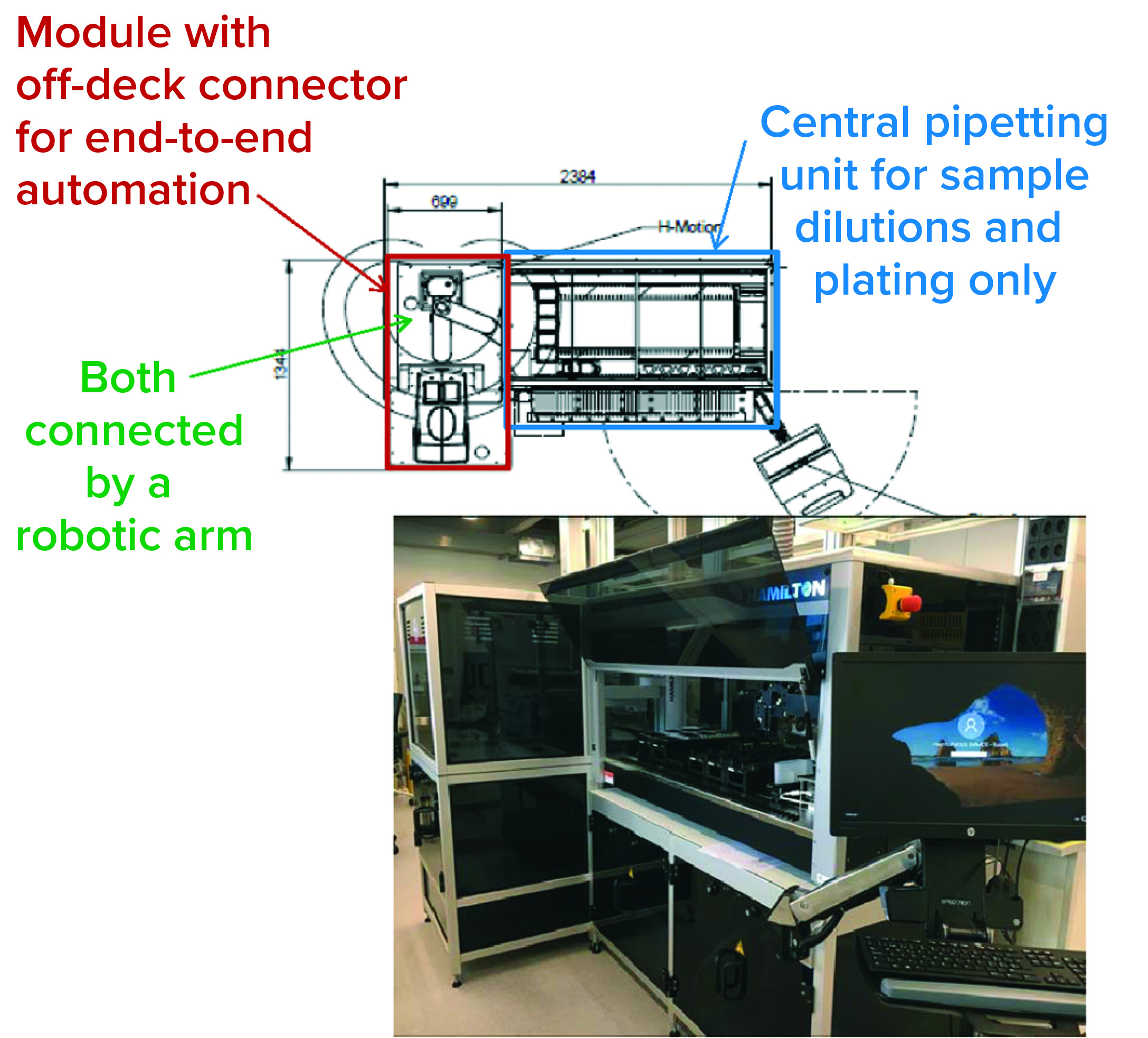
Figure 2: The Roche global bioassay automation team’s approach
Assay Development
Technologies: After choosing the Hamilton Microlab STAR liquid-handling platform, the Roche team defined a standard configuration. That included setting a number of pipetting channels and a configurable deck layout, for example, and detailing the system in a specifications document. Method files are intended to be stored on and transferred through a central server accessible to all Roche bioassay laboratories around the world. “The idea is that you can just run a method from this global-share server.”
Bioassays can be automated to different extents. Low-level automation is just mechanical pipetting (e.g., sample preparation), with the rest of the work done by hand; a high level is full end-to-end automation of every step, including incubation, plate washing, readout, and so on. Most solutions fall between those extremes in a “semiautomated” format. “If you want to do more than just pipetting,” Beck said, “you need to integrate off-deck devices.”
The core of most laboratory robotics systems is a liquid-handling device used for sample dilutions and plating. If more steps of a bioassay than just pipetting are to be automated, then centrifuges, incubators, shakers, plate readers, and so on need to be integrated. “Usually, these are off-deck devices,” Beck explained. “They are not on the pipetting deck and need to be connected by a robotic arm. Ideally, assays can be automated to an extent that they could run overnight without manual interactions. That would be a really big benefit, of course.”
Setting things up requires a great deal of effort. And it’s worth noting that an end-to-end automation approach can monopolize the system for a significant amount of time. “If you’re just pipetting, then it takes just an hour or so before the system is free again.” With that in mind, Beck’s team made flexibility a priority. “Not every assay needs to be end-to-end automated. And even methods that are do not always need to be run under the same conditions.” So the decision whether certain work steps will be manual or automated is made case by case, depending on system use, the number and types of assays to be performed in a given workflow. Free system time can be gained for other, additional assays if required by automating some steps and performing other steps manually.
Figure 2 shows a bioassay automation system consisting of a central pipetting (liquid handling) unit in a defined standard configuration and a module containing “off-deck” stand-alone devices, currently only implemented in the development department. Both parts of the system are connected by a robotic arm for plate transport between the central pipetting unit and different off-deck devices.
In addition to such relatively large systems designed for integrating different work tasks, small benchtop robots (capable only for performing simple pipetting tasks) are used in Roche bioassay laboratories. Such small systems can provide a “helping hand” for sample dilution and plating. Because of their low complexity, small footprint, and relatively low price, those can represent a “low-threshold” automation solution. Accordingly, one or even several such helping hands could be implemented per laboratory relatively easy. “Unfortunately,” Beck said, “at least in our experience, no such small system currently available can do such work with GMP compliance.”

Figure 3: Results from the first automated enzyme-linked immunosorbent assay (ELISA) at Roche were inconsistent, with high variances between automated and manual methods.
Case Study: He offered one example of a technical pitfall encountered in the Roche laboratories. Setting up an automated assay is not simply “plug and play.” This is illustrated by an experience with automating an ELISA in 2012, for example. Figure 3 shows dose-response curves from manual and automated initial runs of the same assay automated (before optimization). They looked very different, with high variability between the plate results from the automated method, which was not acceptable. “So we had to go the ‘stony road’ of troubleshooting to identify the problem and fix it.” Beck says most laboratories can expect such problems early on.
To identify the root cause of a variability problem, the Roche team divides the assay into its component steps, evaluating each one separately rather than repeatedly performing the whole assay. For example, after pipetting fluorescein solution, technicians look for deviations in its signal from certain wells that should have had the same sample concentration. Using this approach, the team quickly discovered variabilities caused by partial aspiration of air in the pipetting process by detecting minimal differences in absolute liquid levels of wells, depending on their position on the plate. Adjusting the immersion depth of the pipette tips ultimately yielded consistent assay results. The laboratory went through 19 revisions of its initial automated method to obtain equivalent results from both manual and automated ELISAs (Figure 4).
“This is a common problem,” Beck said. “In most cases, the reasons for problems observed are quite simple, but it can be a challenge to find the root cause.”
There should be no difference in raw data over long-term trending whether assays were performed by different technicians or by robots. The results should be indistinguishable. That creates and justifies the flexibility to choose based on circumstances. “For example, we want to have the choice of doing things manually if the system is down or blocked, without having to requalify a method.”
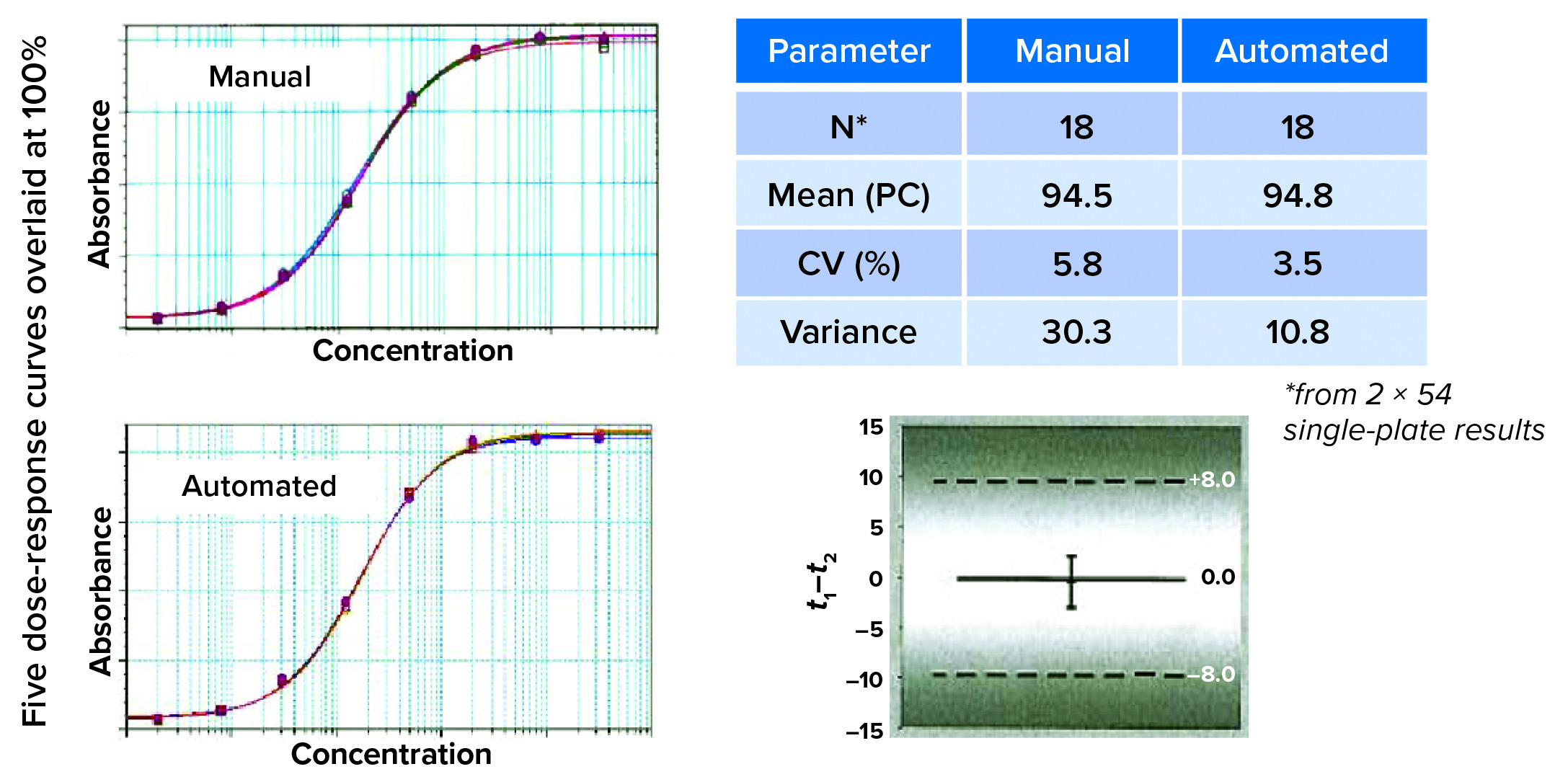
Figure 4: Finding equivalency; on visual inspection, the curves look similar. That must be confirmed by comparing recovery and variant results. Here, the mean of the product control is similar for recovery and assay variance. Statistical evaluation (a one-sided t-test) concluded that automated and manual assay methods were interchangeable.
Validation: That like-for-like concept affects how automation assays are validated. Some people might say, “Our robot is just a big pipette, and we don’t validate pipettes for every product. So why should we do it for our robots?” Views differ greatly here, though. Is a robot “just a big pipette,” or can it be thought of better as “another technician”? The answer influences your validation approach: e.g., automation as a pure robustness parameter or as part of intermediate precision.
“In our experience, it’s hard to set up a method and bring a system online. It requires a lot of knowledge.” But once that effort has been expended for an optimized base method, that can be used as a framework for efficient programming of new methods. The first provides a basic blueprint for those to come. New methods do not need to be programmed from scratch, but rather are copies of a template in which pipette volumes and incubation times are adapted, worksteps can be copied and pasted in from other methods, and so on.
“Potency assays in most cases are quite similar,” said Beck. “So in most cases, it’s sufficient just to adapt, and you might insert or delete some steps. This makes it relatively easy to set up a new method.”
| Requirements of 21 CFR Part 11 |
|
Business Aspects
Computerized systems must comply with 21 CFR Part 11 (2), which is a complex and laborious process, as the “Requirements” box shows. For example, data integrity is mandatory, so an audit trail provided by the system must show changes, manual interactions for error handling, change control of method scripts, and so on.
As for user training, not everyone needs to be an expert. “You need at least two specialists, the method programmers. They have a high level of expertise, they can set up and/or adapt methods, and they have vendor contacts. These people maintain system qualification, develop methods through troubleshooting, and train other users.”
All others work on an operator level, running automated methods. If there’s a problem, they go to the method programmers, who can support several labs or even different sites with their expertise. Anyone can learn relatively quickly how to run automated assays. To reach the level at which you can program a method or at least change or optimize existing methods requires a great deal of training over a stretch of time.
The topic of automation is polarizing, Beck said. When implementing the concept, laboratory staff can exhibit a broad spectrum of motivation, acceptance, reservations, and concerns. Some people are enthusiastic from the start: “I want to be first to work with the robot!” Others may be resistant: “It’s always worked without one. It’s easier for me to just use the pipette.” Some may be fearful: “If all assays are automated, will you need us at all?” Many will take a “wait-and-see” approach.
“I cannot give simple guidance on how to handle such concerns,” Beck said. “It has a lot to do with information. You have to talk with the people about it. And management should not praise automation as a tool for reduction of full-time employees (FTEs); there won’t be less but rather different work to do.”
He highlighted the normal evolution of change, from polarization and skepticism — to acceptance and familiarity — to a sense of “implicitness. At the end, they say they would never go back.” People simply need time to adjust.

Figure 5: New horizons for assay formats; the expected increase in quality and efficiency exceeds what can be accomplished manually.
New Horizons
Automation should enable biopharmaceutical quality laboratories to use more complex assay formats. Figure 5 shows an extreme example for illustration. “You have different dilutions, one after the other, in decreasing concentrations, and so on — all on one microplate. Plate-position effects are quite common, and you prevent them by mixing the positions up a bit. That takes complex pipetting, which is not easy for a human being. A robot can do it easily.”
Another option that automation brings is the 384-well microplate format. That provides for more complex assays and obtaining more data points per run. “We think this [plate format] can increase greatly the quality and efficiency of our assays,” says Beck. But he cautions that it would eliminate the like-for-like concept (the principle that robot and human technicians are somehow interchangeable) when assays can be performed only by robots.
Whether potency-assay automation will benefit an organization depends on many factors that are difficult to calculate simply. Some are specific to certain workflows and organizations. Beck recommends that companies match and adapt the extent of automation to their own specific needs. A modular setup for stepwise implementation and use of automation provides flexibility, and it can reduce risks of implementing too much complexity at once and overburdening laboratory staff. He says that laboratories need to expect technical obstacles and prepare to embark on a steep learning curve. Among staff, some psychological barriers can arise, and automation for GMP compliance is possible but a particular challenge.
Robots can take over work packages to relieve people from exhausting and repetitive tasks, giving laboratory technicians time for work that machines cannot do. And a robot can perform tasks that are too complex for a human to do with reasonable effort and an acceptable error rate. The different aspects described herein might be helpful for readers deciding whether and how to implement laboratory robots for QC bioassays.
References
1 ICH Q2(R1). Validation of Analytical Procedures: Text and Methodology. US Fed. Reg. 62(96) May 1997: 27463–27467; https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf.
2 Electronic Records; Electronic Signatures. 21 Code Fed. Regs. Part 11; https://www.govinfo.gov/app/details/CFR-2011-title21-vol1/CFR-2011-title21-vol1-part11.
Further Reading
Bartolotto E, et al. Assessing Similarity with Parallel-Line and Parallel-Curve Models: Implementing the USP Development/Validation Approach to a Relative Potency Assay. BioProcess Int. 13(6) 2015: 26–37; http://lne.e92.mwp.accessdomain.com/upstream-processing/assays/assessing-similarity-with-parallel-line-and-parallel-curve-models-implementing-the-usp-developmentvalidation-approach-to-a-relative-potency-assay.
Bower KM. Certain Approaches to Understanding Sources of Bioassay Variability. BioProcess Int. 16(10) 2018: S6–S8; http://lne.e92.mwp.accessdomain.com/upstream-processing/assays/certain-approaches-to-understanding-sources-of-bioassay-variability.
Lamerdin J, et al. Accelerating Biologic and Biosimilar Drug Development: Ready-to-Use, Cell-Based Assays for Potency and Lot-Release Testing. BioProcess Int. 14(1) 2016: 36–44; http://lne.e92.mwp.accessdomain.com/upstream-processing/assays/accelerating-biologic-and-biosimilar-drug-development-ready-to-use-cell-based-assays-for-potency-and-lot-release-testing.
Shi J, McKee M. Building a Robust Biological Assay for Potency Measurement. BioProcess Int. 13(4) 2015: 50–55; http://lne.e92.mwp.accessdomain.com/analytical/product-characterization/building-a-robust-bioassay-for-product-potency-measurement.
Hermann Beck, PhD, is principal scientist and leads the bioassay automation team in pharma technical development Europe (biologic) analytics at F. Hoffmann-La Roche Ltd. in Basel, Switzerland; hermann.beck@roche.com. Maribel Rios is managing editor, and Cheryl Scott is senior technical editor for BioProcess International, part of Informa Connect, PO Box 70, Dexter, OR, 97431; cheryl.scott@informa.com.