Potency is a critical quality attribute of a biological product and is often determined by a biological assay (also called bioassay or biopotency assay). Specifically, potency is the biological activity or capacity of a product directly linked to its clinical efficacy. Potency tests are performed as part of product release, comparability studies, and stability testing. Nonbiological methods — which measure a product’s molecular or biochemical characteristics (e.g., ligand-binding assay) — have gained interest as replacements for often troublesome bioassays. Even with recent advancements in alternative methods, regulatory agencies expect manufacturers to make considerable efforts to first develop a bioassay, largely because of the method’s ability to directly assess a product’s biological function. The results of a bioassay can be used to gauge manufacturing consistency and product shelf life.
Whether based on cell culture, tissue models, or animals, bioassays are inherently variable because a living system is used. Many factors can affect the outcome. For example, results from a cell-based bioassay can be influenced by cell bank conditions, cell thawing, cell passage, culture medium, cell maintenance methods, and cell manipulation during the assay (just to highlight a few parameters).
Regulatory agencies and industry experts have provided guidance on bioassay development and validation. The ICH Q6B guideline recommends that bioassays measure an organism’s biological response to a product, a biochemical or physiological response at the cellular level, enzymatic reaction rates, or ligand- and receptor-binding (1). The FDA issued a guidance for industry on potency tests for cellular and gene therapy products in 2011 (2). Three recently revised USP chapters on biological assay development and design offer detailed recommendations on experimental design, statistical analysis, and assay validation (3–5). And several bioassay forums allow scientists to express their views of the role of bioassays in lot-release and stability testing and appropriate assay acceptance criteria (6, 7).
An important aspect of bioassay design and development is to ensure that the final assay is robust enough to measure manufacturing consistency and is correlated with clinical outcomes. To achieve that, a bioassay requires continuous development and refinement throughout a biologic’s life cycle. In many cases, developing a matrix of R&D assays at preclinical and early clinical stages is beneficial. As products and assays are better understood, one or more relevant assays can be selected for further optimization. Ultimately, those assays will be validated, and product specifications will be set based on their performance. Figure 1 shows the recommended time frame for developing and validating a suitable potency assay during a product development life cycle. Generally, assay validation should be complete before or early in phase 3 so that the assay is ready for release and stability testing of a licensed product.
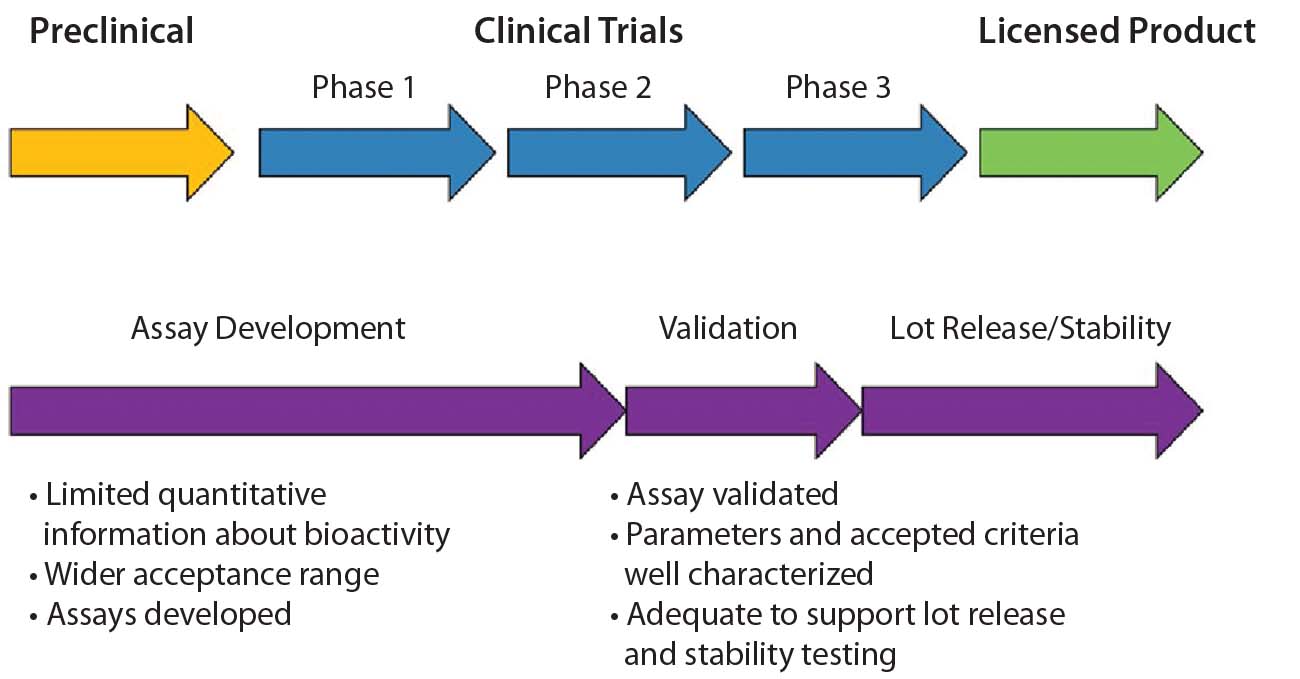
Figure 1: Time frame for potency assay development and validation
Here we focus on potential aspects to consider when building a consistent cell- based potency bioassay that will be suitable as a current good manufacturing practice (CGMP) release test.
Adequate Cell Type or Cell Line
To develop a cell-based potency assay, first determine which cells are appropriate. If possible, select a type that is relevant to a product’s mechanism of action (MoA) and is known to respond well to the product. For instance, when developing a monoclonal antibody (MAb) that binds to a cancer cell marker and subsequently leads to growth inhibition of target cells, screen several malignant cell lines that express that marker. The most responsive cell line should be selected.
Unless required, primary cells should not be used because of their potential for lot-to-lot, donor-to-donor variability. However, in some cases where primary cells must be used, consider appropriate approaches to minimize cell heterogeneity. That can be done by securing a large lot of cells or isolating a subpopulation when feasible.
Peripheral blood mononucleated cells (PBMCs) are commonly used in bioassays for products with hematopoietic indications. But PBMCs lack consistency in potency tests in general, primarily because only a subset of cells generate the response of interest. Furthermore, the percentage and activity of different subpopulations of PBMCs vary from run to run and between lots. Instead of running a potency assay using cryopreserved PBMC populations, and when the assay allows, isolate a desired cell population and use the “purer” cells in a biopotency assay.
Second, characterize the cell line on which the potency assay is based. Information about cloning history, genetic stability, growth characteristics, and passage limits all should be established. At minimum, evaluate passage limits and vial-to-vial consistency in the potency assay. In addition, create and store phase- appropriate cell banks. It is not unusual to use a research-grade cell bank for early phase potency assay development and performance. However, when a product progresses to phase 2–3, it is critical that you make and characterize well a cell bank generated under a more controlled (e.g., CGMP) laboratory environment. Whenever a new bank is generated — in addition to the standard purity and identity testing — test cells from that new bank in the assay to ensure that the assay parameters are comparable with the current bank.
Last, but not the least important, ensure that cells are in the necessary physiological state and behave in the potency assay as expected. For suspension cells, establish the minimum and maximum cell density for culture maintenance. Do not under- or overtrypsinize adherent cells. The former is likely to cause unwanted cell selection, and the latter can potentially damage the cell membrane. Cells should not be allowed to grow over confluent to prevent potential cell transformation.
Material Consistency and Stability
Reference Standards: Well-defined and controlled materials and reagents are essential elements for a successful bioassay for product potency. The design of these assays and calculation of relative potency for a product rely heavily on reference standards. Selecting the right material to serve as the reference standard is important. Some companies use a sample from an R&D process as the reference in an early stage potency assay (when the assay itself most likely is also under development). However, as an assay development progresses, select an appropriate reference standard early in the process.
The biological response of a test sample is directly compared against the reference standard in a potency assay. So the reference standard is ideally generated from a similar manufacturing process as the test sample and with known stability data under intended storage conditions. Moreover, the reference should be evaluated thoroughly through multiple runs in the potency assay (n > 10) to establish a “normal” range for EC50 (the half maximal effective concentration), hill slope, and upper and lower asymptotes when the assay uses a 4-PL or 5-PL data- fitting model commonly used for potency assay evaluation. When the reference is deemed appropriate for a given assay, allocate sufficient quantities of material for future assays. It is likely that the material will be used not only for assay development and validation, but also for sample testing when its shelf life allows. When the current lot is close to depletion, retain some samples for use in a bridging study to compare with the new reference standard.
The reference standard is not the only critical reagent in potency assays. Often, other reagents have a strong impact in assay outcomes. Although they may differ from assay to assay, common critical reagents for biopotency assays include serum for cell culture or reaction medium, enzymes, transduction/transfection reagents, and primary and secondary antibodies. During assay development, evaluate multiple lots of critical reagents. It is not unusual for a specific lot to work better than another for a potency assay. When that happens, make every effort to secure as much material as possible, taking shelf lives and testing volumes into consideration. When a reagent is stored frozen and thawed before use in an assay, prepare aliquots to prevent multiple freeze–thaw cycles. As with reference standards, new lots of critical reagents require bridging/qualification studies before implementation in well-controlled, validated assays.
Procedural Accuracy
A robust potency assay requires precise and accurate procedures. This also applies to nonbiological analytical assays such as an enzyme-linked immunosorbent assay (ELISA). More important (because of the inherent, nonrobust nature of biopotency assays) is the use of well-defined and accurate procedures. From a stock solution, both the reference standard and test sample are diluted over multiple steps to the final working dilution (concentration) range tested in the assay. In addition, a potency bioassay involves pipetting cell suspension to one or more 96-well microplates and mixing with other reagents. Without accurate pipetting, there is no solid foundation for a robust potency assay.
We do not discuss pipetting techniques at length here, but rather offer a few quick points to consider. First, work with a volume that is close to each pipette’s calibration volume. Second, use prewet tips to increase consistency. Third, except for cell suspension, all reagents should be at room temperature for accurate pipetting. Last, use reverse pipetting when dealing with viscous liquids.
Incubation temperature and time should be well controlled. By contrast with an assay performed in an R&D environment, a regulated assay must have a well-defined range for acceptable incubation temperature and time. Many good laboratory practice (GLP) or GMP laboratories have incubation chambers (incubator, refrigerator, or freezer) for 37 °C, refrigerated, or frozen conditions but no chambers for room temperature. As a result, plates are placed on the bench top for room-temperature incubation. This “room temperature” can range from 20 to 35 °C, even 15–40 °C. Fluctuations across the range of temperatures can significantly affect assay outcomes. For incubation steps that are performed at room temperature, using an incubator set at 20–25 °C can reduce assay variability. As for incubation time, do not use a wide range of times for critical incubation steps, if possible. For example, a 60 ± 10 minutes time window is much better than one to two hours.
Consistent washing steps are essential for controlling assay background and precision between replicate wells. Whether using manual washing or an automated plate washer, be consistent and allow only one washing step method in the procedure. When an assay requires manual washing, ensure that all analysts wash plates in a similar way — working through the plate at the same orientation, adding wash buffer at similar speed, and washing adjacent rows at similar intervals. When using a plate washer, make sure the same setting is used every time.
Proper and timely calibration and maintenance of equipment also can contribute to procedural accuracy. All equipment used in GMP assays should be validated for their intended use.
Analyst Training
For an assay that is not completely automated, the analyst is the largest source of assay variability. This is especially true for a biopotency assay that involves multiple dilution steps and manipulation of test sample, cells, and reagents. Onsite training can be conducted when transferring an assay to a different laboratory. This training provides the personnel from the originating and the receiving laboratories an opportunity to observe each other. Cross-training allows analysts to identify steps that might not be documented in an assay’s standard operating procedures but are important to assay performance. On many occasions, the “transferer” (the laboratory that is more familiar with the assay) can provide information about equipment or reagents that differ between the originating and receiving laboratories.
When an assay is performed infrequently, a periodic requalification program can familiarize analysts with assays and prevent potential assay failure due to long gaps between assay performance. The frequency of requalification depends on the complexity of the assay and the proficiency of the analyst. Generally, if an analyst has not run a given assay for six months, a requalification run should be performed before performing a GMP release test.
Data Analysis
The design of a bioassay that reports a relative potency value for a test sample against the reference standard takes into account run-to-run variability to some degree. Some assays are still highly variable despite thorough evaluations of the sources of variability. That is possibly attributed to the wide and unpredictable biological response being measured. For such assays, averaging final potency results from two or three independent setups or runs can be a useful approach to reduce the risk of the assay results being influenced by random factors. This strategy has been adopted by many scientists developing biopotency assays, especially for effector assays such as an antibody-dependent cell-mediated cytotoxicity (ADCC) assay or a complement-dependent cytotoxicity (CDC) assay. In such cases, the assay is qualified or validated based on two or three runs, the same as described in governing documents.
Troubleshooting a Nonrobust Bioassay
The above discussion provides several key aspects that could affect biopotency assay robustness. Below are some approaches for troubleshooting a nonrobust bioassay.
Dissecting a complex bioassay to individual steps is sometimes very helpful. When the response from cells plated in a 96-well microplate is measured after incubation with a number of reagents, evaluate the response after each step, if possible, to identify the problematic step in the procedure. Starting from a base plate with cells only often provides some clues such as position/edge effect or uneven cell seeding or growth.
A design of experiments (DoE) study is a useful tool to evaluate multiple variables systemically. You can perform DoE at the assay development stage to identify optimal assay conditions or for assay troubleshooting. For example, reagent concentrations, incubation time, and cell density all can be incorporated into a one DoE, rather than be part of separate evaluations. DoE enables assessment of the impact from related experimental conditions that cannot be achieved by changing variables one at a time.
Data trending should be implemented to monitor performance of a biopotency assay. Key factors that could potentially negatively affect assay outcomes, such as operator, cell lot number, passage number, material lots, and equipment identification should be recorded. Other assay parameters such as EC50 values, hill slopes, and upper to lower asymptote ratios can also be trended. Those data often can answer questions such as
- What has changed from when the assay was running well?
- Is there a trend?
- What is the most likely root cause for the assay failure?
Data trending also helps detect data shift or drift before a system suitability failure or out-of-specification event. Once a trend has been identified, preventative actions should be taken to prevent assay failure.
Setting limits goes hand-in-hand with data trending. For example, when a trend shows that an assay does not work well once cells have been cultured for more than 20 passages, then set a cell passage limit in that assay protocol. Knowing method limits such as cell passages, specific reagent lots, and analyst-specific parameters is valuable and helps exclude potential factors that could introduce variability.
Keys to Success
A successful bioassay suitable for validation and final-product lot release may take multistage development and fine-tuning to reach a final design. Although many roadblocks can present on the way to a robust bioassay, controlling variables at early stage assay development and careful quality control in assay performance are key to a meaningful potency test to ensure product quality.
References
1 ICH Q6B: Test Procedures and Acceptance Criteria for Biotechnological/ Biological Products. US Fed. Reg. 64, 1999: 44928.
2 Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. Food and Drug Administration: Rockville, MD, 2011.
3 USP Chapter <1032> Design and Development of Biological Assays. US Pharmacopeial Convention: Rockville, MD. 2012.
4 USP Chapter <1033> Biological Assay Validation. US Pharmacopeial Convention: Rockville, MD, 2012.
5 USP Chapter <1034> Analysis of Biological Assays. US Pharmacopeial Convention: Rockville, MD, 2012.
6 Rieder N, et al. The Roles of Bioactivity Assays in Lot Release and Stability Testing. Bioprocess Int. 8(6) 2010: 33–42.
7 Robinson CJ, et al. Assay Acceptance Criteria for Multiwell-Plate-Based Biological Potency Assays. BioProcess Int. 12(1) 2014: 30–41.
Jing Shi, PhD (shijing_sj@hotmail.com) was senior scientist and head of the Immunoassay Laboratory at BioReliance. Corresponding author Marian L. McKee, PhD is director, of US Development Services at BioReliance, 14920 Broschart Road, Rockville, MD 20850; marian. mckee@bioreliance.com.