A space-filling model of allantoin (HTTP://STOCK.ADOBE.COM)
Endotoxin contamination has been the bane of the bioprocessing industry since its inception. Endotoxins are everywhere: They are toxic and/or interfere with every type of therapeutic, diagnostic, and research product; they are indestructible within the limits of product tolerance; and they are difficult to remove (1–4). Beyond that, they interact with various biological species in ways that prevent accurate measurement (5, 6).
Managing these issues has been a focus of the industry for at least half a century, yet it still too often falls short of manufacturing controls necessary to satisfy regulators (7, 8). It is even more of a problem with diagnostic and research products because their preparation generally involves fewer controls. Development of new endotoxin removal methods thus remains a subject of persistent interest.
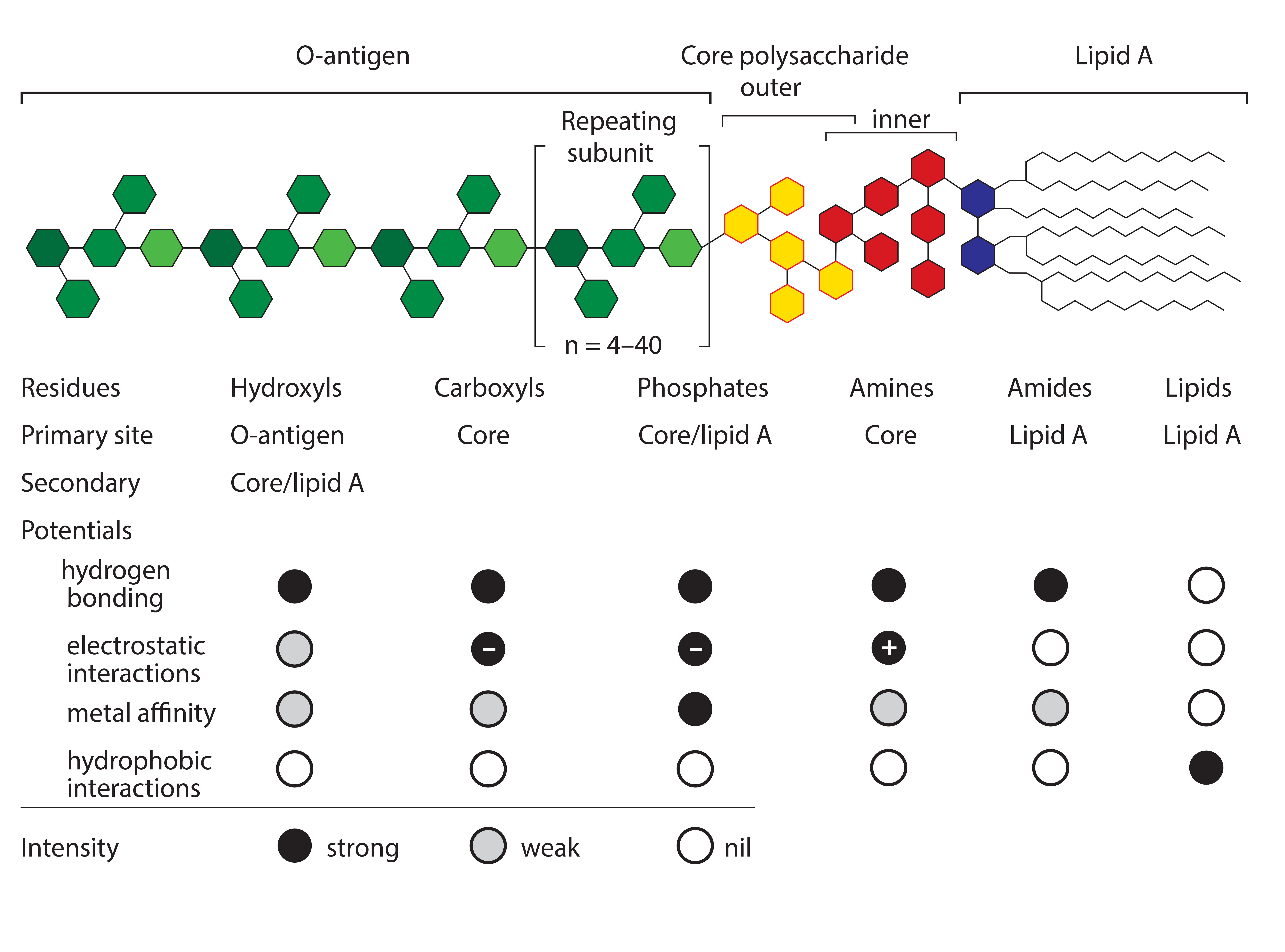
Figure 1: Endotoxin basic structure and chemical potentials
Endotoxin removal methods typically focus on particular regions of an endotoxin molecule. Positively charged chromatography media historically have dominated the field. Examples include traditional anion exchangers and a number of multimodal electropositive adsorbents such as histidine, polymixin B, and Capto Adhere and Toyopearl NH2-750F media (9–13). These methods focus on the negatively charged oxygen atoms associated with the carboxyl and phosphoryl residues of the core polysaccharide and lipid A regions of the molecule (Figure 1).
In many cases, multimodals support more effective endotoxin removal than traditional anion exchangers, but they have two limitations: They bind many proteins, and their effectivity is dramatically altered by variations in pH and conductivity. These limitations have their least impact with alkaline proteins such as IgG1 monoclonal antibodies that flow through columns at mildly alkaline pH and low conductivity. Endotoxins bind well under such conditions. Increasing conductivity or reducing pH to enable antibody flow-through tends to compromise endotoxin binding in parallel.
Biological affinity chromatography with a recombinant version of the protein that enables the Limulus amoebocyte lysate (LAL) test also exploits the core polysaccharide and lipid A regions (14) of endotoxins. It bears the same conveniences of other biological ligands: exquisite selectivity, no need for sample equilibration, and minimal development burden. But it also shares their burdens: elevated expense and intolerance of rigorous sanitization conditions.
The metal affinity of the core polysaccharide and lipid A phosphoryl groups can be exploited with either hydroxyapatite (HA) or immobilized metal affinity chromatography (IMAC). Endotoxin phosphoryl residues have especially high affinity for the calcium component of hydroxyapatite (15). They also bind iron, aluminum, copper, and zinc ions, among other metal ions (16). Proteins that elute from HA in chloride salts at low phosphate concentration support the best endotoxin removal. IMAC is more complex due to the necessity of selecting and binding metals in addition to the usual tasks of method development, but it also tolerates high salt concentrations.
Hydrophobic-interaction chromatography might be expected to target the hydrophobicity of the O-and N-linked fatty acids of the lipid A region, but it has not proven competitive. The high salt conditions required for protein binding copromote formation of stable self-associations among endotoxins that make the lipid A region inaccessible. The only method that effectively exploits hydrophobicity of lipid A at present involves addition of the Triton X114 surfactant (17). Incubation at room temperature to promote association with lipid A is followed by refrigeration that causes the detergent to gelatinize, facilitating its removal with still-associated endotoxin.
All these methods leave the largest and most exposed part of the endotoxin molecule unexploited: the O-antigen, also known as the O-polysaccharide. It consists of repeating groups of sugars that can extend up to 100 nm from the core polysaccharide. For reference, this is eight times the longest dimension of IgG. The O-antigen generally has no charged residues, no residues by which metal affinity ligands might interact, and no hydrophobic residues, but it is highly enriched in hydrogen donors and acceptors.
The O-antigen behaves in aqueous solutions like the hydrophilic tail of a surfactant whereas the lipid A region behaves like the hydrophobic head. Endotoxin associations lack the symmetry of surfactant micelles because of their complex coreactions with metal ions, proteins, and other species. Instead, they produce ribbons, sheets, and less ordered structures into the millions of Daltons, but the net result remains the same: They restrict steric access to the core polysaccharide and lipid A regions while they enhance exposure of the O-antigen.
This article discusses two new approaches to endotoxin removal. One describes a methodological enhancement to anion-exchange chromatography that eliminates the need for sample equilibration. Traditionally, it exploits the negatively charged residues of the core polysaccharide and lipid A regions. The other method uses a new adsorbent that exploits hydrogen bonding with the O-antigen. Its selectivity and capacity are unaffected by pH, conductivity, chelating agents, surfactants, and organic solvents. These approaches are procedurally complementary in addition to being chemically complementary, so they offer powerful combined endotoxin reduction.
Overcoming Dependency of Anion Exchange on Sample Composition
Two modes of operating ion exchangers are well known: bind-elute and flow-through. Both terms refer to the behavior of the product of interest. Both modes require advance equilibration of a sample. A third operational mode represents a special case of flow-through mode: void exclusion (18). It fractionates on the basis of size in addition to charge interactions, and it does not require sample equilibration.
Anion exchange in void-exclusion mode (VEAX) limits sample volume per cycle to the same volume as the column void, the space between the particles. This corresponds to about 40% of a gravity-packed bed. Proteins with the same charge as the exchanger are repelled and unable to enter any of the particle pores, so they travel exclusively through the void volume and elute first. IgG is a good example. Unbound but nonexcluded proteins diffuse into and out of particle pores. This gives them a longer flow path and causes them to travel through the column at a slower rate than proteins that travel exclusively through the void.
Buffers and salts flow more slowly than unbound proteins because they have access to pores that are too small for proteins. As with buffer-exclusion chromatography on size-exclusion media, salts exit the column after the void fraction. This is important because it liberates VEAX users from the need to equilibrate a sample before chromatography. Samples eluted from bioaffinity columns at extreme pH can be applied to VEAX without adjustment. Samples at 2.0 M NaCl from a multimodal cation exchanger can be applied without adjustment. Samples under physiological conditions – for example, those drawn directly from cell culture – can be applied without adjustment. VEAX works even with samples applied in 1 M guanidine.
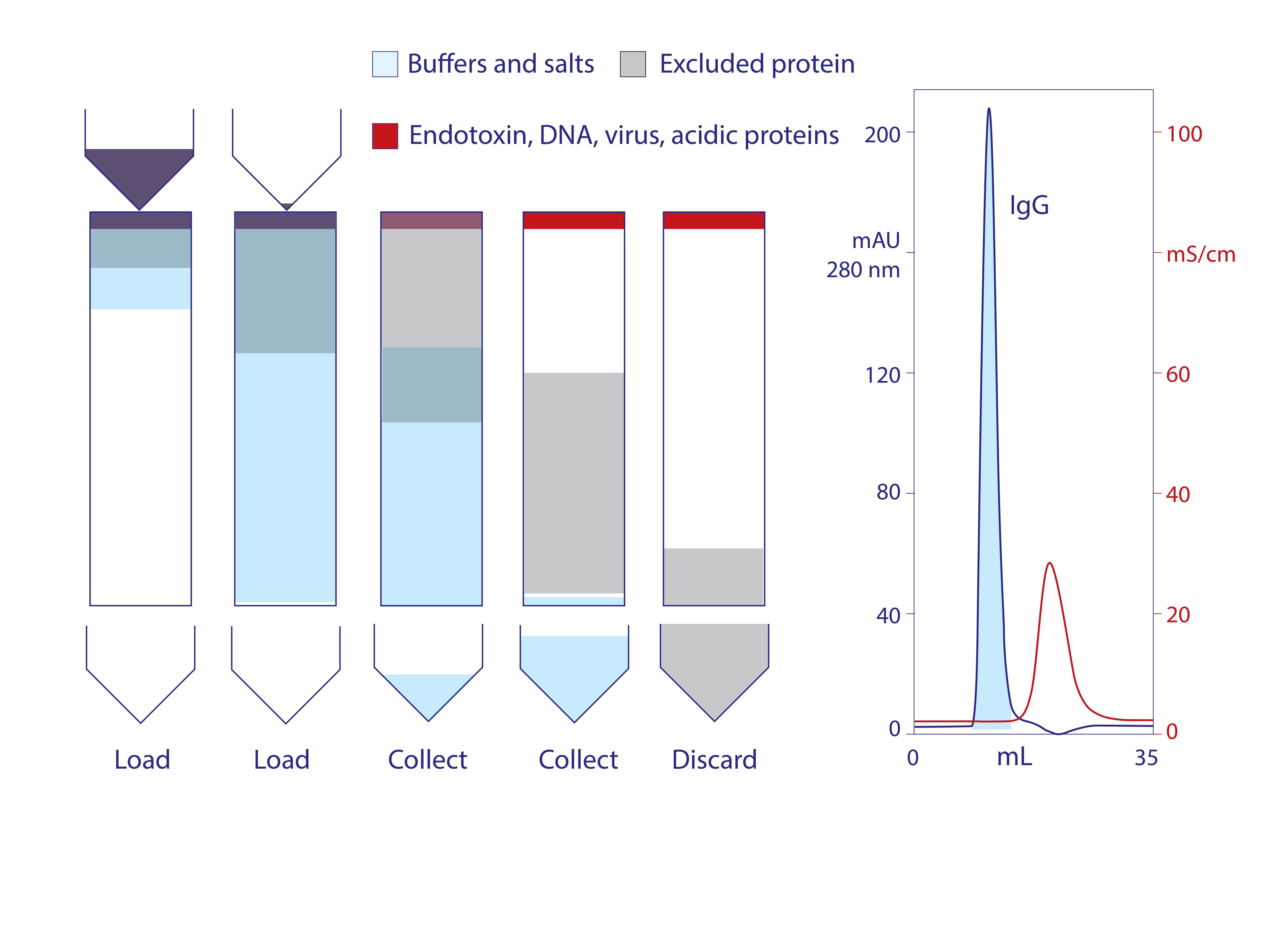
Figure 2: Stages of anion exchange in void-exclusion mode (VEAX) and VEAX of protein A−purified human monoclonal IgG1; chromatogram redrawn from (18)
Endotoxins, DNA, virus, and acidic proteins, if present, bind strongly and can mostly be eluted later with a pulse of 1–2 M NaCl. Cleaning with 1 M NaOH removes any stubborn remnant. Figure 2 diagrams VEAX fractionation and shows an example with protein A purified IgG. Figure 3 illustrates chromatograms of VEAX performed on an IgG1containing cell culture harvest, with analytical size-exclusion chromatography (SEC) profiles before and after VEAX. VEAX also works well with VHH, Fab, and F(ab′)2 (Figure 4).
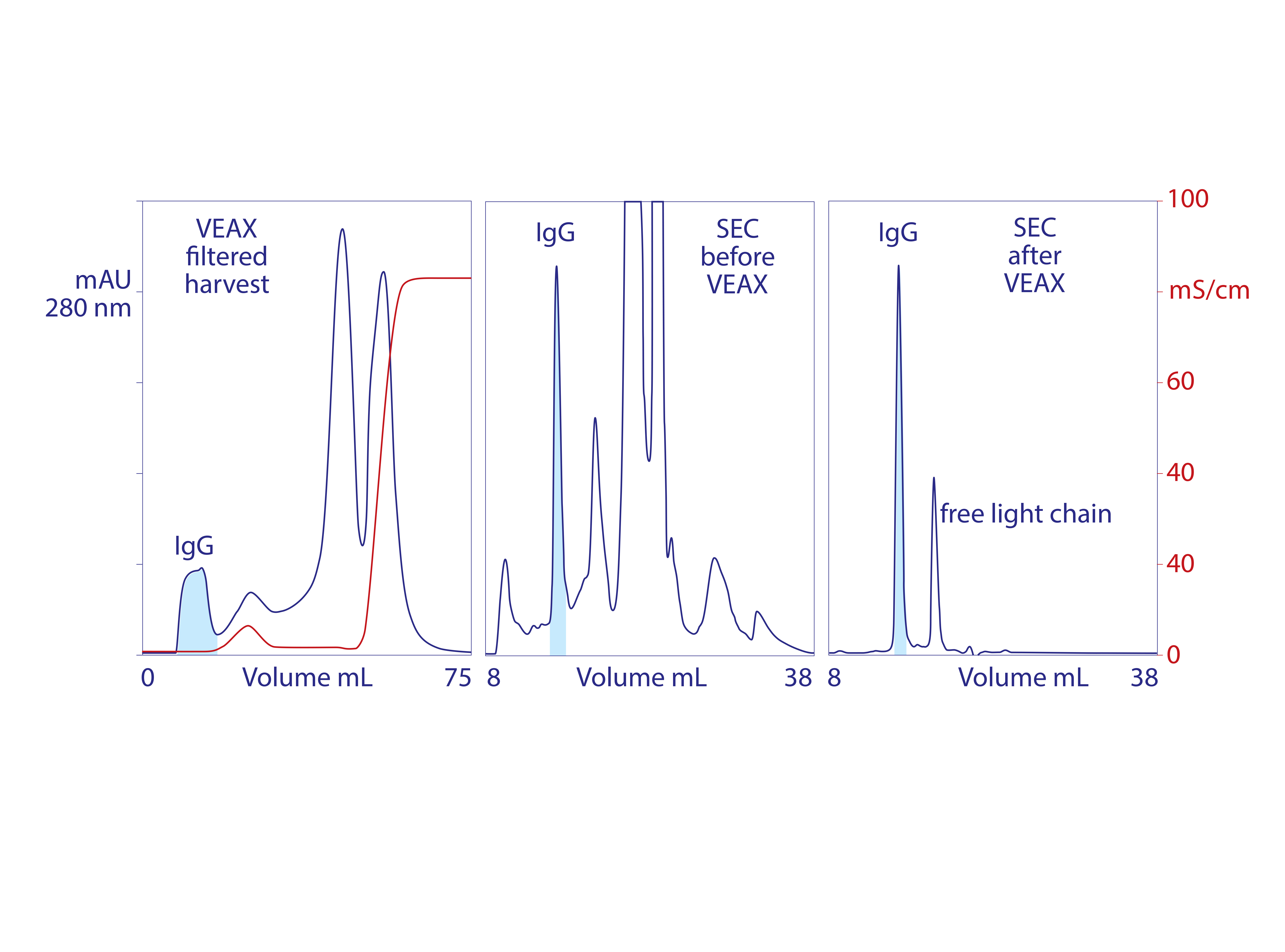
Figure 3: VEAX of monoclonal IgG1 from cell culture harvest with analytical size-exclusion chromatography (SEC) profiles; chromatograms redrawn from (18)
Beyond simplifying workflows, unrestricted tolerance of sample composition has special importance for proteins that form stable associations with endotoxins. It enables sample pretreatment with one or more dissociative additives. As a sample advances through the column, product liberated by the dissociating agent(s) of interest is excluded to the void volume. Some endotoxin that was previously associated with the product typically elutes with the dissociating agent(s). Otherwise, it remains bound to the exchanger.
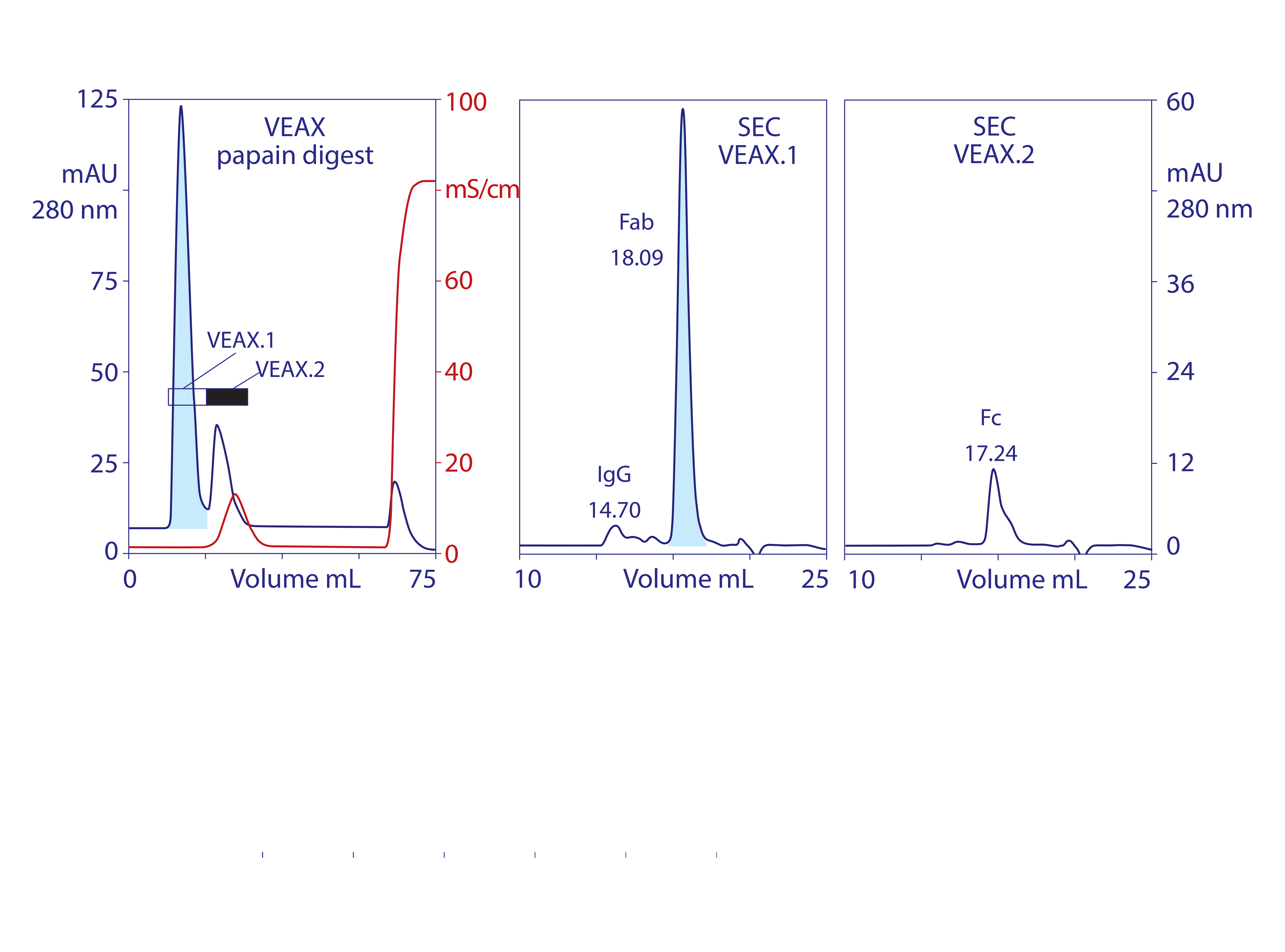
Figure 4: VEAX of a papain digest to product Fab, with analytical SEC profiles; chromatograms redrawn from (18)
The dissociating agent(s) can be any small molecule: urea, arginine, guanidine, sodium chloride, organic solvents, surfactants, or combinations at any concentrations necessary to release a product from its association with an endotoxin (19). But this raises the question: Why doesn’t the protein reassociate with the endotoxin at the void front as dissociating agents are left behind? Reassociation is thought to be blocked competitively by the anion exchanger. Dissociated endotoxins at the void front are attracted more strongly to the massively electropositive chromatography surface than they are to the comparatively less electropositive proteins excluded to the void volume.
Development of column conditions is simple. Start by equilibrating the column in the absence of salt at the highest pH at which the product of interest elutes completely within the bounds of the void. A good place to start with IgG is 20 mM Tris, pH 8.5. If antibody does not elute in the void fraction, then lower the pH. If pH 6.5 does not permit exclusion, then perform design of experiments (DoE) to identify the highest pH and lowest salt concentration that will permit void exclusion of the product. If indications suggest that the target protein might be stably associated with endotoxin, a convenient shortcut is to add 1 M guanidine to the sample. Dissociative conditions tailored to the particular target protein can be developed later.
Some older generation anion exchangers do not work for VEAX, but many introduced over the past 20 years do (18). With some antibodies, void exclusion may be achievable with multimodal anion exchangers. Expect this to enhance the degree of endotoxin removal, but such media will be applicable to fewer proteins and under a narrower range of conditions than are traditional anion exchangers.
VEAX removes 2–4 logs of endotoxin, DNA, and viruses, and it also achieves substantial antibody purification. Aggregates are highly reduced, and the only significant impurity class is excess light chain (Figure 3). Similar results are achieved with Fab, F(ab′)2, VHH (Figure 4), and generally with alkaline peptides and proteins; however, do not rely on isoelectric point (pI) as a guide. Charge distribution is more important. An IgM with a pI of 9.1 failed to elute even at pH 6.0 because negative charges localized on its Fc regions caused it to bind the anion exchanger (18).
O-Antigen Binding Through Hydrogen Bonding
Identifying a solid phase with ability to bind solutes exclusively by hydrogen bonding is a challenge because the ligand has to compete with all the hydrogen bonding occurring among water molecules. Nature provides some guidance in the form of nucleotide base pairs that maintain integrity of DNA by hydrogen bonding despite being solubilized in an aqueous environment. Hydrogen bonding between as few as four base pairs is enough to maintain stability. The next level of the challenge is to obtain a sufficiently high surface density of hydrogen donors/acceptors to enable high capacity adsorption. Nature provides guidance here as well: crystals.
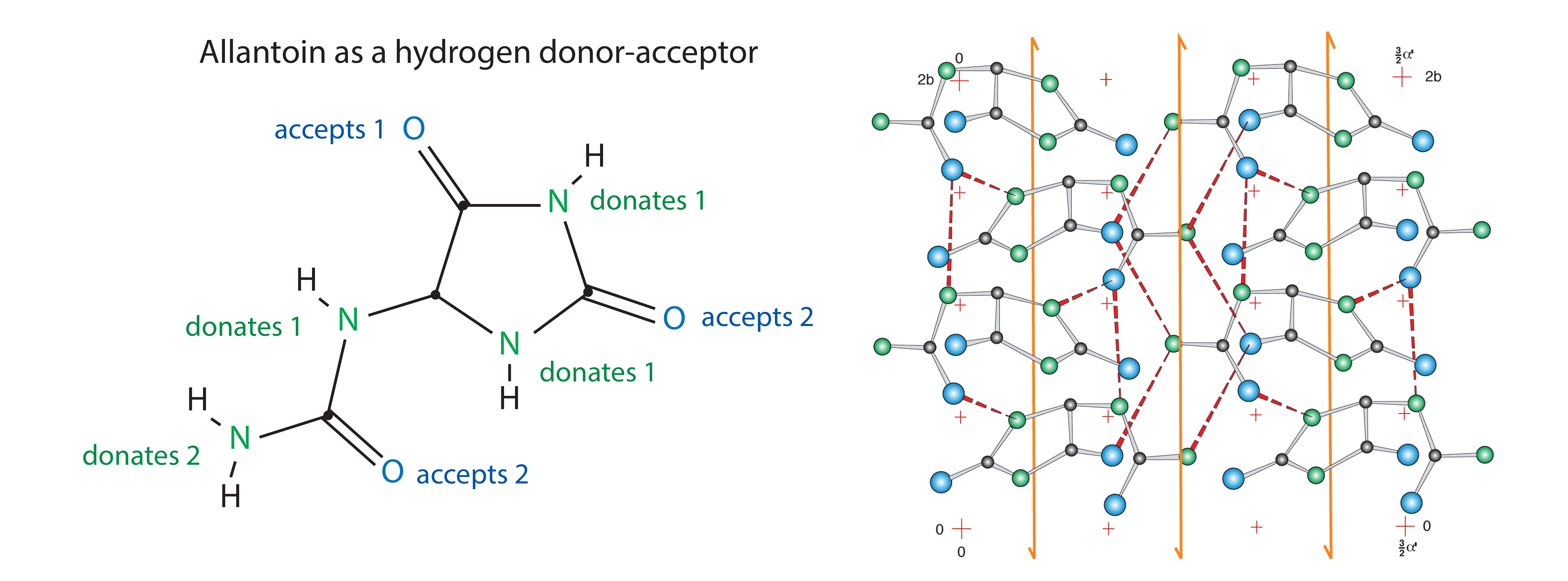
Figure 5: Allantoin structure and crystal surface; structures redrawn from Mootz (29)
All these conditions are met by a naturally occurring compound known as allantoin: (2,5-dioxo-4-imidazolidiny) urea. Allantoin is a nonionic eukaryotic metabolite formed by oxidation of uric acid during the breakdown of DNA (Figure 5). It occurs at trace concentrations as a soluble intermediate in human physiology. It forms 500-nm cuboid crystals at concentrations above 0.56%. These crystals exhibit picomolar affinity for endotoxins (Ka = 1.010 M-1 under physiological conditions — higher than for all known antiendotoxic peptides, endotoxin-binding proteins, and other engineered endotoxin adsorbents (20–22). Binding capacity is estimated at 80 million endotoxin units per gram of crystals.
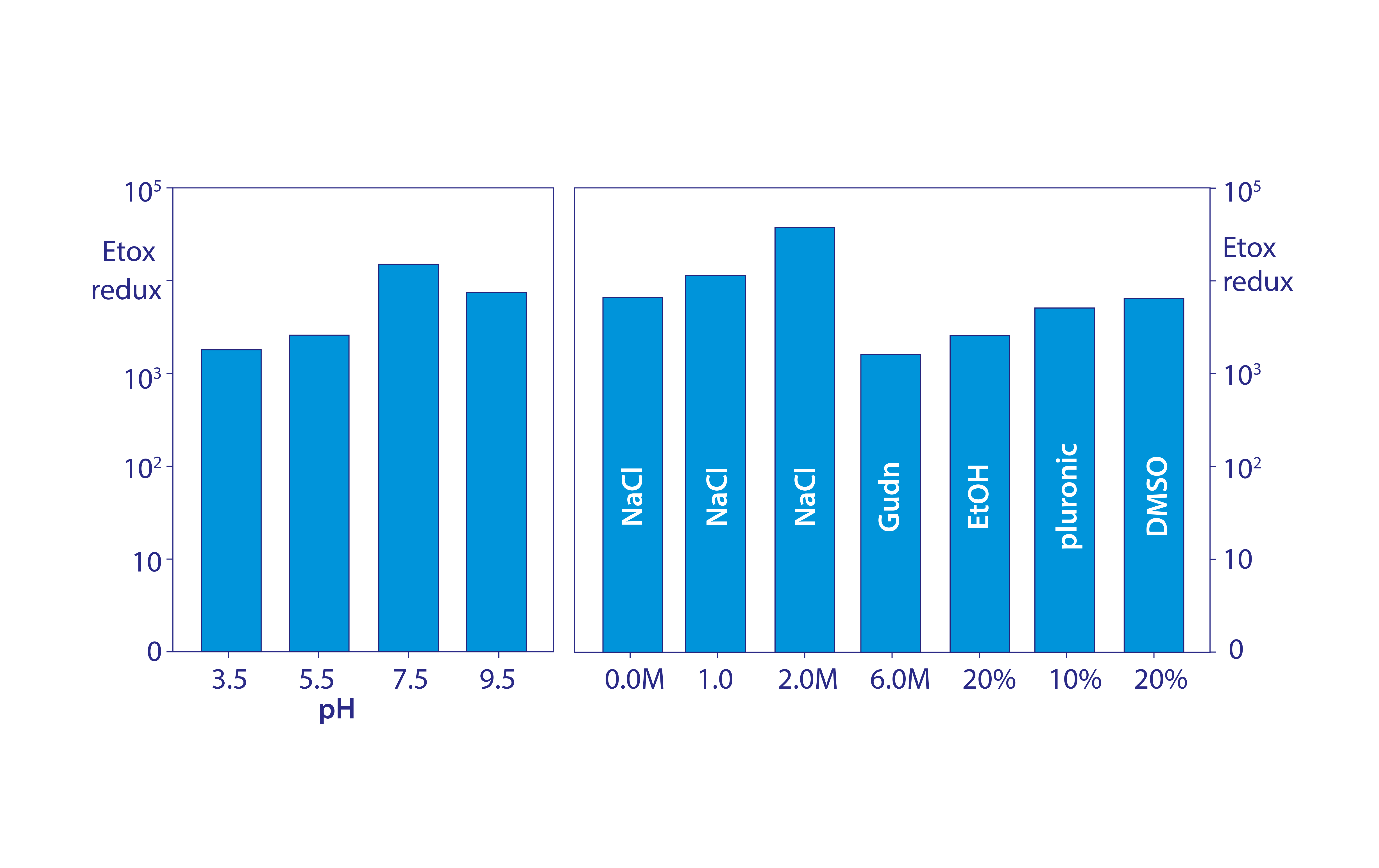
Figure 7: Conservation of endotoxin binding by allantoin crystals in different chemical environments; data replotted from (18, 20)
Figure 7 shows that allantoin crystals maintain 2–4 logs of endotoxin removal from pH 3.5 to 9.5. Endotoxin removal also tolerates the presence of 20% glycerol, 20% ethanol, 20% dimethyl sulfoxide (DMSO), 10% detergent, 2 M NaCl, and even 6 M guanidine. These findings eliminate metal affinity, electrostatic, and hydrophobic interactions from the possible mechanism of binding. Urea, diazolidinyl urea, and formamide — all amide-containing compounds — competitively blocked endotoxin binding. Amides are uncharged but effective hydrogen donors.
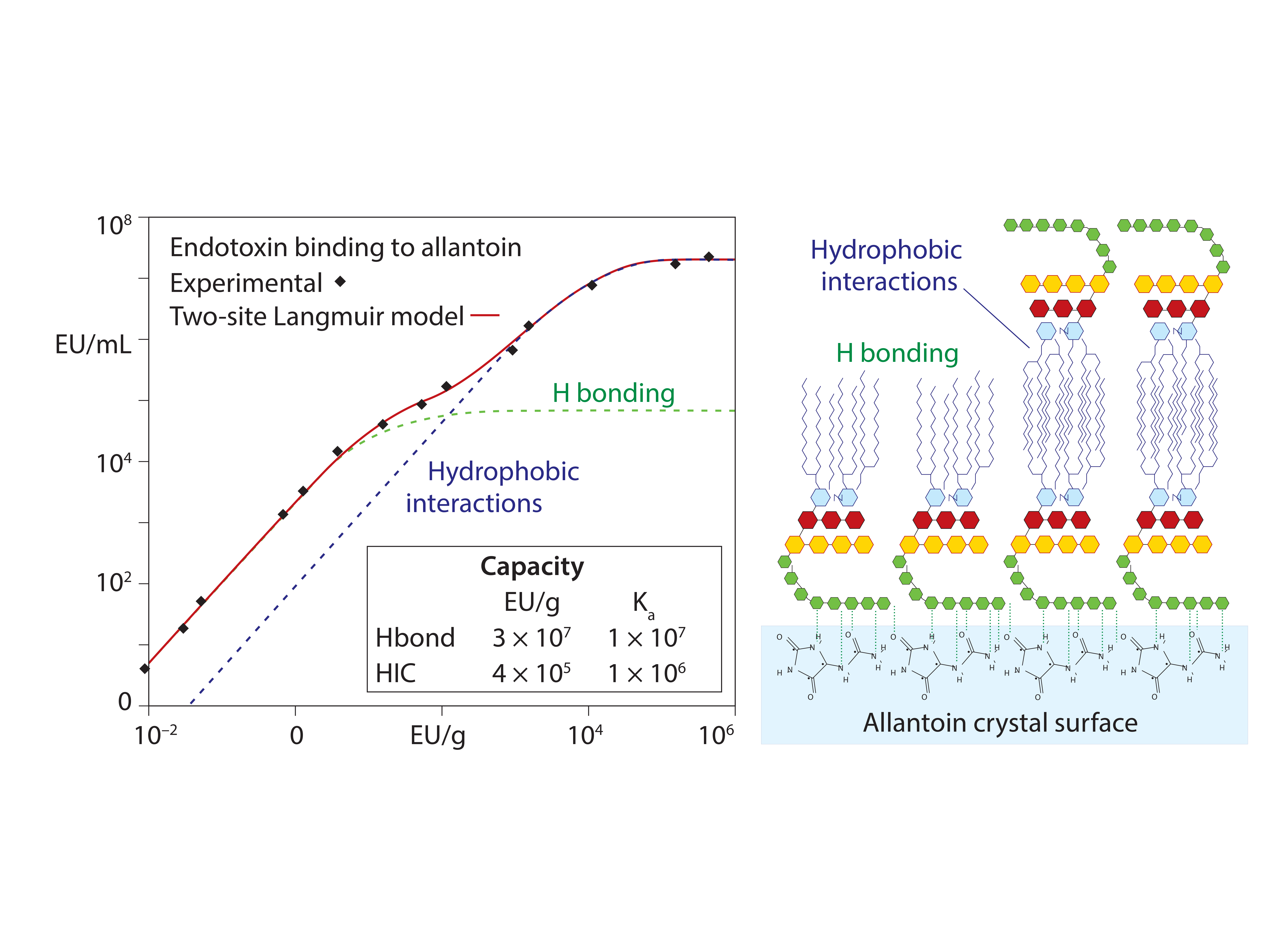
Figure 6: Allantoin binding to endotoxin: Langmuir isotherm and conceptual model; redrawn from (20)
These results support the conclusion that endotoxin binding is mediated through exposed amide residues on allantoin crystal surfaces. Allantoin binding sites on endotoxins are dominated by the sugars of the O-antigen. However, carboxyls, phosphates, and free electron pairs on ethanolaminyl nitrogen residues (if present) enable hydrogen bonding by the core polysaccharide and lipid-A regions (Figure 1). Experimental data further indicate accumulation of endotoxin double layers on allantoin crystal surfaces. Figure 6 illustrates a hypothesis consistent with these data: The O-antigen region is bound to the allantoin crystal surface, exposing its lipid-A region to the solvent, thereby creating a strongly hydrophobic substrate for secondary binding to the lipid-A regions of other endotoxin molecules.
Binding capacity for proteins smaller than 100 kDa is nil and contributes to high recovery, regardless of whether the protein is acidic or alkaline, hydrophobic or hydrophilic. Losses increase with protein size but remain moderate at allantoin concentrations of 10% or less. Figure 4 shows the relationship between endotoxin reduction and protein recovery for polyclonal IgG. On the plus side, allantoin’s affinity for larger assemblages also manifests in disproportionate aggregate removal and reduction of virus. Dengue, influenza virus, and phage are reduced by 3 logs (23, 24).
Method development requires nothing more than adding allantoin crystals to the target protein solution. Start with a default of 10%. If losses are excessive for large proteins, consider checking lower concentrations. Reaction kinetics essentially are instantaneous, probably because of the high affinity constant. Remove the endotoxin-loaded crystals by sedimentation or filtration. Select the concentration that achieves the best balance of endotoxin reduction and protein recovery. If indications suggest product binding to endotoxin, evaluate addition of dissociating compounds in combination with allantoin.
Allantoin has yet to become established in production of research, diagnostic, and therapeutic recombinant proteins, but it comes to the field with a solid pedigree. Toxicity concerns are allayed by its being a normal human metabolite. It is widely accepted by regulators worldwide as an additive to over-the-counter drugs, shampoos, toothpastes, and skin care products. Industrial quantities are produced exclusively through synthetic processes. It is USP listed (25), and several high-sensitivity assays have been published for documenting its removal from IgG-containing solutions and purification process intermediates (26–28).
Recommended Practice
A key benefit of using allantoin instead of other endotoxin-removal methods is its versatility. Because it is not limited by ionic or hydrophobic interactions, it can be applied to any protein regardless of its pI or hydrophobicity. Its parallel tolerance of wide-ranging chemical conditions eliminates the need for sample preparation and offers the ability to dissociate endotoxins from proteins that bind them. Its ability to remove 3 logs of virus simultaneously is another bonus. In fact, its range of operating conditions overlaps with common virus-inactivation methods, offering an opportunity to combine allantoin coprecipitation with low pH or solvent-detergent inactivation — an ability that ultimately could prove as valuable as endotoxin removal.
For alkaline proteins, combining allantoin coprecipitation with VEAX offers extraordinary capabilities where ultra-low endotoxin levels may be needed; for example, where there may be gross endotoxin contamination or where endotoxins may be stably associated with the target protein. VEAX offers the same ease of sample preparation and tolerance of dissociating conditions as allantoin but adds 3 logs of DNA removal and compounds the ability of allantoin to remove endotoxins, viruses, and aggregates. The allantoin step meanwhile facilitates VEAX by removing contaminants that might foul or consume the endotoxin-binding capacity of an anion exchanger.
These capabilities recommend the combination of allantoin and VEAX as a routine platform approach for purification of low-endotoxin research antibodies. At laboratory scale, a follow-on SEC step can remove enough of the remaining contaminants to meet standards for in vivo use. The sample-loading capacity of SEC is limited, but it requires no sample equilibration, it requires virtually no method development, and it offers the critical feature of supporting rigorous NaOH treatment. Precleaning the SEC column with NaOH ensures that endotoxins do not return to a sample from which they have previously been removed.
For commercial-scale purification of research and diagnostic antibodies, allantoin paired with VEAX also can be used in place of protein A or protein G. It involves a larger number of cycles but fewer buffers and no expensive chromatography media. The follow-on SEC step can be replaced by faster-throughput, higher-capacity methods such as multimodal anion exchange, cation exchange, or HA to remove of free light chain. In the simpler case of removing endotoxins from already-purified proteins, allantoin, VEAX, or both can be applied according to the opportunity and needs of the situation.
Acknowledgments
Thanks to the Bioprocessing Technology Institute in Singapore, where allantoin coprecipitation and VEAX were developed originally under a grant from the Singaporean Agency for Science, Technology and Research (A*STAR).
References
1 Ongkudon J, et al. Chromatographic Removal of Endotoxins: A Bioprocess Engineer’s Perspective. ISRN Chromatogr. 2012 Article ID: 649746.
2 Karkar A, Rees A. Influence of Endotoxin Contamination on Anti-GBM Antibody Induced Glomerular Injury in Rats. Kidney Intl. 52(6) 1997: 1579–1583.
3 Watanabe J, et al. Endotoxin Contamination of Ovalbumin Suppresses Murine Immunologic Responses and Development of Airway Hyper-Reactivity. J. Biol. Chem. 278(43) 2003: 42361–42368.
4 Woods J, et al. Resistance to Meningococcemia Apparently Conferred By Anti-H.8 Monoclonal Antibody Is Due to Contaminating Endotoxin and Not to Specific Immunoprotection. Infect. Immun. 55(8) 1987: 1927–1928.
5 Bryant R, et al. Endotoxin Contamination of Enzyme Conjugates Used in Enzyme Linked Immunoadsorbent Assays. J. Clin. Microbiol. 17(6) 1983: 1050–1053.
6 US Food and Drug Administration. Guidance for Industry: Pyrogen and Endotoxin testing (2012); www.fda.gov/downloads/DrugsregulatoryInformation/Guidances/UCM310098.pdf.
7 Lolas A. Microbial Control Strategies in Bioprocessing Falling Short of Assuring Product Quality and Satisfying Regulatory Expectations. American Pharm. Rev. (2013); www.americanpharmaceuticalreview.com/Featured-Articles/134040-Microbial-Control-Strategies-in-Bioprocessing-Falling-Short-of-Assuring-Product-Quality-and-Satisfying-Regulatory-Expectations.
8 Suvarna K, et al. Case Studies of Microbiological Contamination in Biological Product Manufacturing. American. Pharm. Rev. 14(1) 2011: 50–56.
9 Issekutz A. Removal of Gram-Negative Endotoxin from Solutions By Affinity Chromatography. J. Immunol. Met. 61(3) 1983: 275–281.
10 Anspach F. Endotoxin Removal By Affinity Sorbents. J. Biochem. Biophys. 449(1−3) 2001: 665–681.
11 Guo W, et al. Removal of Endotoxin from Aqueous By Affinity Membrane. Biomed. Chromatogr. 11(3) 1997: 164–166.
12 Chen H, et al. Factors Affecting Endotoxin Removal from Recombinant Therapeutic Proteins By Anion Exchange Chromatography. Prot. Expr. Purif. 64(1) 2009: 76–81.
13 Hirayama C, et al. Cross-linked N,NDimethylaminopropylacrylamide Spherical Particles for Selective Removal of Endotoxin. J. Chromatogr. A 676(2) 1994: 267–275.
14 Hoess A, Lidington R. Lipopolysaccharide-Binding and Neutralizing Peptides. World Patent WO 1995005393 A2 1993l; www.google.com/patents/WO1995005393A2?cl=en:Method.
15 Gagnon P, et al. A Ceramic Hydroxyapatite-Based Purification Platform: Simultaneous Removal of Leached Protein A, Aggregates, DNA, and Endotoxins from MAbs. BioProcess Intl. 4(2) 2010: 50–60.
16 Tan L, et al. Differential Interactions of Plasmid DNA, RNA, and Endotoxins with Immobilized and Free Metal Ions. J. Chromatogr. A 1141(2) 2006: 226–234.
17 Aida Y, Pabst M. Removal of Endotoxin from Protein Solutions By Phase Separation Using Triton X-114. J. Immunol. Met. 132(2) 1990: 191–195.
18 Nian R, et al. Void Exclusion of Antibodies By Grafted-Ligand Porous Particle Anion Exchangers. J. Chromatogr. A 1282 (2013): 127–132.
19 Ritzen U, et al. Endotoxin I Monoclonal Antibody Preparation Using Arginine. J. Chromatogr. B 856(1−2) 2007: 343–347.
20 Vagenende V, et al. Amide-Mediated Hydrogen Bonding at Organic Crystal/Water Interfaces Enables Selective Endotoxin Binding with Picomolar Affinity. ACS Appl. Mat. Interfaces 5(10) 2013: 4472–4478.
21 Vagenende V, et al. Self-Assembly of Lipopolysaccharide Layers on Allantoin Crystals. Colloids Surf. B Biointerfaces 120 (2014): 8–14; doi: 10.1016/j.colsurfb.2014.04.008.
22 Vagenende V, et al. Allantoin As a Solid Phase Adsorbent for Removing Endotoxins. J. Chromatogr. A 1310 (2013): 15–20; doi: 10.1016/j.colsurfb.2014.04.008.
23 Gagnon P. Selective Binding of Biological Targets to Solid Phase Ureides. United States Patent US 9637724 B2; www.google.com/patents/US9637724.
24 Gagnon P, Vagenende, V. Materials and Methods for Removing Endotoxins from Protein Preparations. United States Patent US 9695216 B2, www.google.com/patents/US9695216.
25 USP Monographs. Allantoin; www.pharmacopeia.cn/v29240/usp29nf24s0_m1400. html.
26 Nian R, Gagnon P. Advance Chromatin Extraction Enhances Performance and Productivity of Cation Exchange Chromatography-Based Capture of Immunoglobulin G Monoclonal Antibodies. J. Chromatogr. A 1453 (2016): 54–61 doi: 10.1016/j.chroma.2016.05.029.
27 Turner R, Stamp L, Kettle A. Detection of Allantoin in Clinical Samples Using Hydrophilic Liquid Chromatography with Stable Isotope Dilution Negative Ion Tandem Mass Spectroscopy. J. Chromatogr. B 891–892 (2012): 85–89; doi: 10.1016/j.jchromb.2012.02.009.
28 Braga R, et al. Development and Validation of a Method for Allantoin Determination in Liposomes and Pharmaceutical Formulations. Spectrochim. Acta part A: Molec. Biomol. Spectr. 91 (2012): 389–394; doi: 10.1016/j.saa.2012.02.022.
29 Mootz D. The Crystal Structure of dl-Allantoin. Acta Cryst. 19(5) 1965: 726–734.
Pete Gagnon is chief scientific officer at Validated Biosystems and member of the BPI Editorial Advisory Board; pete_gagnon@mac.com.
Great article and very useful. It seems the figure illustrating the relationship between allantoin and poly IgG recovery is missing. Would like to see it! Thank you.