
2,000-L stainless steel bioreactor
As an increasing number of cell therapies move into late-phase trials, developers are considering innovative solutions to address scale-up and commercialization challenges. Many of their questions focus on the technologies and engineering strategies that will be needed to optimize their processes, especially bioreactors.
At the January 2016 Phacilitate Cell and Gene Therapy World conference, Siddharth Gupta, a scientist at Lonza (Walkersville, MD), talked about the effects of upstream process decisions on product quality in his presentation “Bioreactor Manufacturing Platforms: So You Think You Know What It Takes, But Do You Really?”
Difficulties and Risks
What are the essential components of developing cell therapies? The 19th century was all about chemistry, and the 20th century was all about physics. Many industry experts are anticipating that this century will be all about biology and its intersection with technology. The only way to make that work is to have great biologists come together with great engineers.
Cell therapy manufacturers should think about the “QCQ” terminology: quality, cost, and quantity. It is important to have cells with the qualities that you need, including phenotype and potency. You should also be able to produce the quantity that you need. Researchers are targeting diseases with extremely large patient populations, such as stroke, congestive heart failure, and different types of blood cancers, all of which require treating thousands of patients every year. So quantity is extremely important. Finally, at every scale — from academic laboratories to late-phase clinical trials — it is important to keep costs in mind. A process must be robust, but it also must give you a final product at a cost that the market can bear.
Why are cell therapy manufacturers having difficulties achieving those three aspects? So far, it is due to the level of process complexity. For example, a car has on average about 15,000 moving parts. But you can touch each part and know how it interacts and function with every other. So if we replace the motor with a better motor, or a gear with a better gear, we know exactly how that affects the car.
By contrast, consider one of the building blocks of a cell, the protein, and we have about 18,000 of them. The problem is that we don’t always know how a protein interacts with other proteins. Our knowledge of protein–protein networks is far from complete. Small changes in a manufacturing environment can greatly affect a final product.
This unknown complexity can lead to unintentional consequences when trying to engineer the cell manufacturing environment. These unintentional consequences have two main causes: obliviousness or overconfidence. Fortunately, not many scientists or engineers find themselves being oblivious, which means intentionally or unintentionally ignoring the complexity of a cell process while making a process change (such as operating pH or operating temperature). In almost all cases, that leads to a change in potency.
By contrast, more manufacturers suffer from overconfidence, either because they rely solely on past experience or because they generally do not understand the effects of small changes. For example, an operator might change pH by 0.5 or dissolved oxygen levels by 5% because it makes the process less expensive or because such a change did not affect a different cell type in the past. But that kind of overconfidence and lack of appreciation for process complexity could lead to a product change and a loss in potency, even though they were made with the goal of process improvement. Just being aware of such unintentional consequences is one step toward a more robust process.
How can manufacturers reduce unintentional consequences? A process engineer must keep three main points in focus:
- If you’re trying to scale up, you first need to implement the right engineering strategies that are not only cost-effective and scalable, but that also don’t affect the biology.
- If a process change has biological effects, they must be minimized to the extent possible.
- You must understand exactly how each of your critical process parameters affect cell identity and potency.
How do companies choose an appropriate process platform? Unfortunately, manufacturers often get caught in a “Catch 22.” For example, for a small, early stage pharmaceutical company developing a cell therapy process, time and money are big constraints. Many companies in these situations decide on a laboratory platform for early stage process development without optimizing their processes. One example of a laboratory-scale process is growing mesenchymal stem cells (MSCs) for allogeneic therapy in T flasks. That is an attractive option in some cases because it requires less time and money upfront. However, as those companies reach phase 2 and phase 3, they need more process changes to meet commercialization needs. There is just no way to produce a commercial allogeneic MSC product in T flasks or in cell stacks. So those companies can run into unintentional consequences when scaling up for commercialization.
The alternative is using a commercially relevant, scaled-up platform from the very beginning. For the MSC example, a commercially relevant platform would be a small bioreactor. But this is not easy. It requires more time, extensive training on how to operate bioreactors (which can be complex), and more funds. Even small bioreactor setup has a much higher initial capital requirement than cell stack or T-flask processes. However, in the long run, the small bioreactors option requires fewer process changes to meet commercialization needs. That is because a commercial bioreactor is simply a scaled-up version of a small bioreactor that you might have used for your phase 1 studies. This option also has fewer unintended consequences and ideally the product behaves exactly the way that it did for phase 1, so you do not need to carry out large comparability studies.
The Catch-22 situation can be resolved in both cases (laboratory platform or scaled-down commercially relevant platform). If you choose the laboratory platform, you need to better understand your processes and the complexity of your cell products. If you use a commercially relevant, scaled-up platform, your option to reduce time and funds is either to partner up with a contract manufacturing organization (CMO) or contract development organization (CDO) or to use a developed off-the-shelf platform. Most CMOs have their version of a platform that can be customized at short notice for a variety of cell therapy products. This option minimizes unintended consequences and reduces upfront time and cost.
What should a company look for when choosing a partner whose platform it can use? Good process development expertise is a must. The CDO/CMO should know how to design experiments and have the technical skill set to conduct your process development. The CDO/CMO partner should have an excellent analytics group to verify that your product does what it’s supposed to do. It should have a dedicated raw material supply chain and excellent regulatory and quality management systems in place. And the CMO/CDO partner should have those commercial-ready platforms for use with your cells.
What platforms are used at Lonza? For allogeneic therapies, Lonza uses stirred-tank, single-use bioreactors. We’ve demonstrated scale-up capabilities from 500 mL to 50 L, and we can consistently grow over 2 million hMSCs/mL, both in 50-L bioreactors and at smaller scales. For our media optimization, we use the ambr 15 system (from Sartorius Stedim Biotech) for growing MSCs on microcarriers. As a result of those studies, we have media for high-density hMSC culture.
For induced pluripotent stem (iPS) cells, we have proprietary animal-free L7 platforms for iPS generation and proliferation. This system consists of a matrix, culture media, and passaging solution. We are currently evaluating the platform for suspension culture of iPS cells.
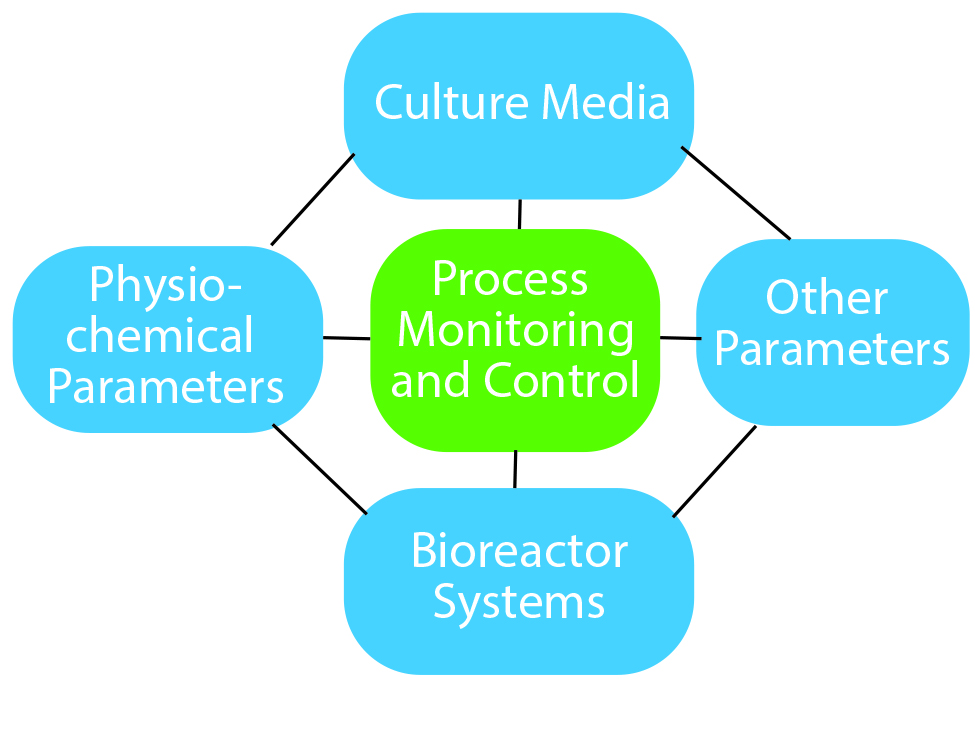
Figure 1: Essential components of process optimization
How does process optimization play a role? It is useful to think of process optimization as a diamond-shaped approach (Figure 1).
On the top is culture media. Here you need to look at not only what goes into media such as the carbon source, amino acids, and vitamins, but also the media feeding strategy. Media components and feeding strategies can affect the cell quality and final product cost.
The second factor includes physicochemical parameters such as pH, temperature, and agitation.
The third part of process optimization is the bioreactor system. The number of single-use systems available for stem-cell culture is quite high. Each of them offers advantages and disadvantages for specific cell types, so you need to evaluate which option offers the best benefits for your cell type and microcarrier type.
The fourth factor covers other parameters, which include — especially for allogeneic therapies — the type of carrier you are growing your cells on. Evaluate a few different carriers on the market and decide which works best for your cell type.
The central component holding everything together is process monitoring and control. You need to ensure that you are using the right process analytical strategy to gain an in-depth understanding of your process. Real-time monitoring of critical process parameters (CPPs) is extremely important, and the industry now has specialty sensors for glucose, lactate, and even cell density. Using such technologies can help you understand your process and provide information in case something deviates from normal behavior.
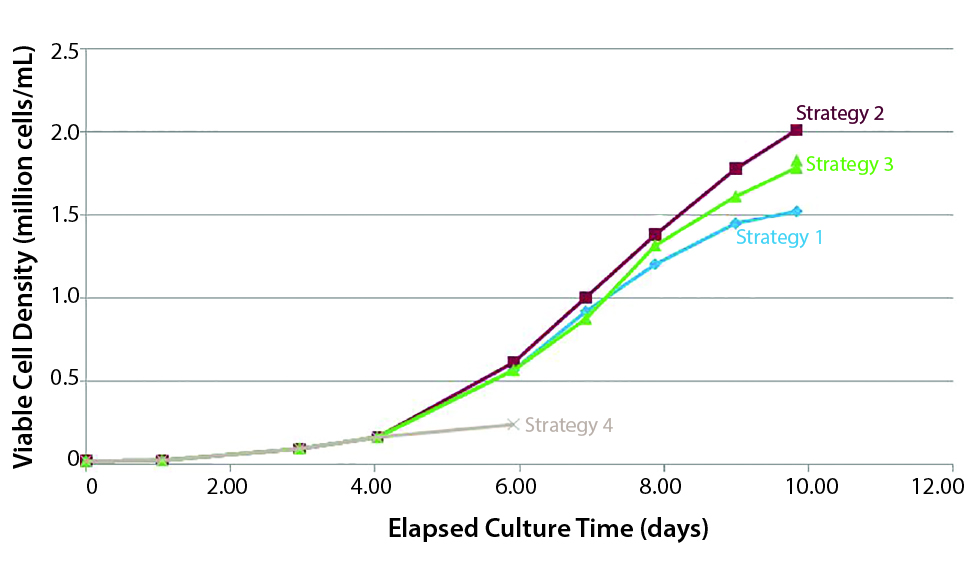
Figure 2: Cell yield dependence on medium feeding strategy (Example 1)
Case Studies
Would you review some examples that show how process changes had unintentional consequences on drug products? Figure 2 shows an example of how cell yield depends on medium-feeding strategy. It shows four media feed strategies for MSCs on microcarriers. The one that yielded the least cells (strategy 4) is a batch feed, in which you start with a certain amount of medium and don’t change it. Because MSCs have a short doubling time, you don’t get very good cell expansion.
Strategy 1 and 2 used the same amount of medium in terms of volume, but the latter performed about 30% better. That is because strategy 1 is a batch-exchange approach, in which you must stop the culture, let all the carriers settle, remove some waste medium from the top, and then add the same amount of fresh medium. That is a labor-intensive process, but many groups are using this strategy.
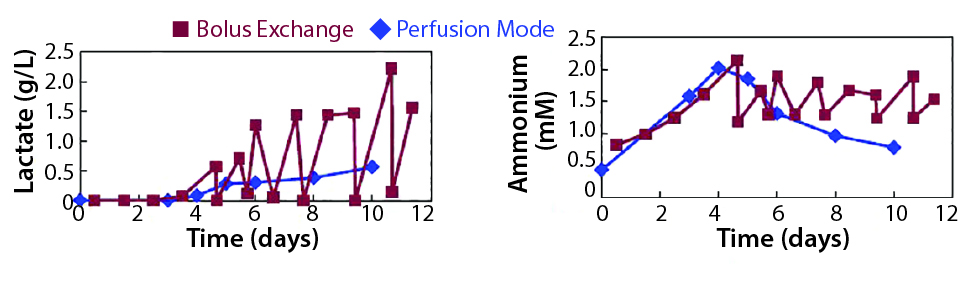
Figure 3: Requirement of optimal medium feeding (Example 1)
Strategy 2 uses the same amount of medium as strategy 1, but medium is exchanged continuously — like perfusion. At the end of culture (day 10) this strategy produced 30% more cells. This difference in cell yield could have multiple causes, but one contributing factor was the accumulation of lactate and ammonia for batch exchange. Figure 3 shows the lactate and ammonia spike up and down for the batch exchange. That happens because between media exchanges, levels of lactate and ammonia build up. When medium is exchanged, levels drop. For perfusion mode, the lactate and ammonia profiles fluctuate less and never reach the levels that might be detrimental to cell growth. So with less labor, you’re able to achieve more cells per milliliter.
In this case, the conventional approach is to settle carriers, but the unintended consequence of using such a manual-heavy process is the build-up of by-products. The solution here is to test multiple-feed strategies — not only perfusion, but also concentrated feed or other novel methods to achieve more cells with less labor.
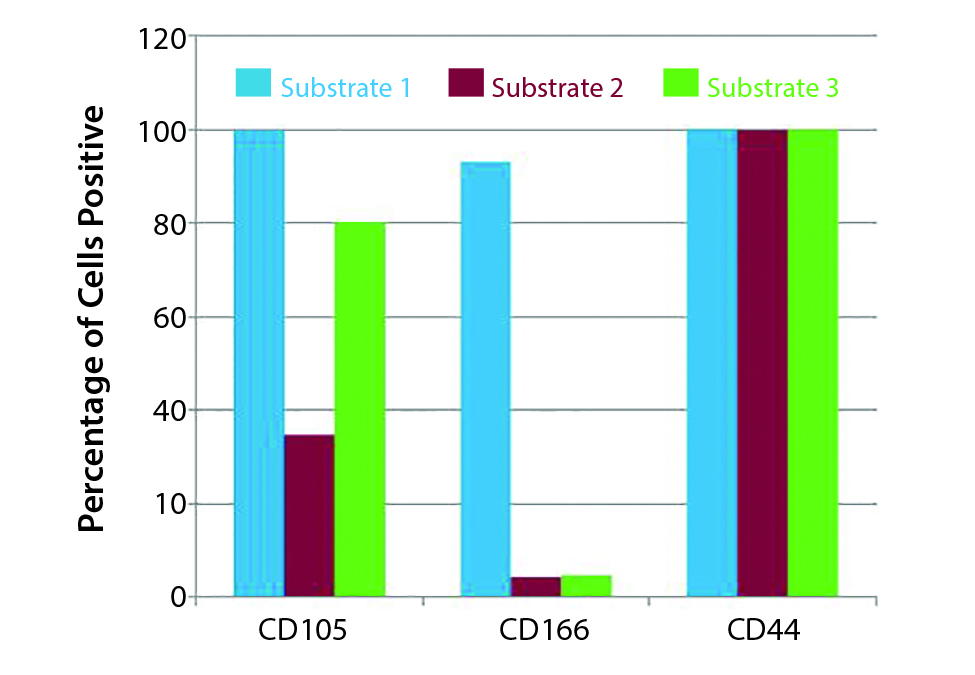
Figure 4: Effect of substrate on hMSC phenotype (Example 2)
Figure 4 shows an example of the effect of substrate on hMSC culture. Here the substrate can be the coating on microcarriers or what those carriers are made of. The conventional approach has been to grow MSCs on polystyrene flasks. We tested three different substrates for hMSC culture. At the end of culture, the percentage of cells positive for CD105 and CD166 were different, but the percentage for CD44 was unaffected.
To prevent such changes, don’t just select the substrate that works in two-dimensional systems because a 3-D environment for cells may be different. Evaluate substrates you think might be used at commercial scale at scaled-down levels. When you go to commercial scale, you will know which substrate works well for your process.
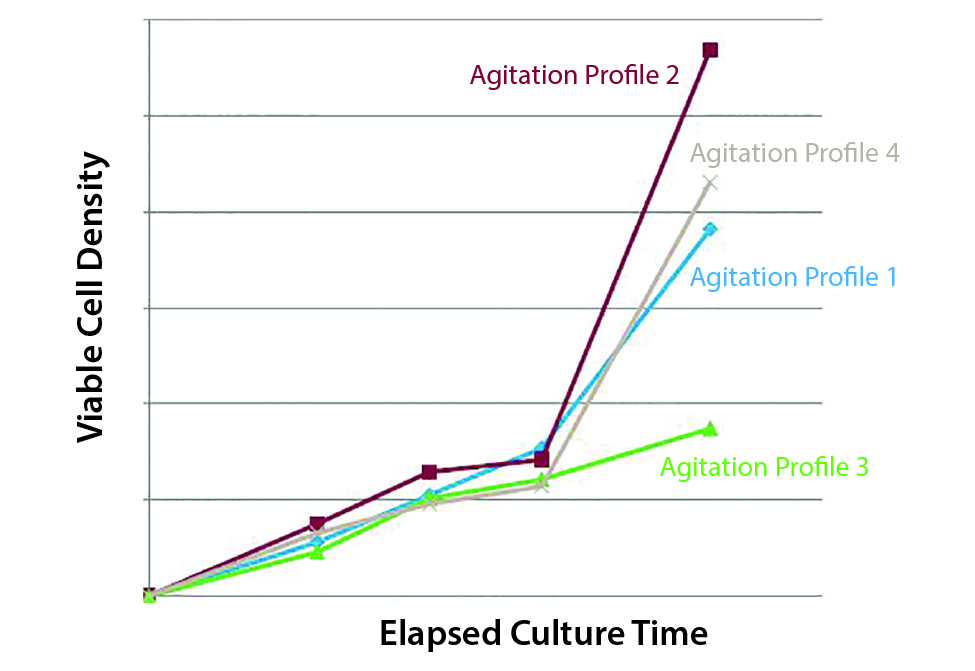 The third example is how the stirring rate affects the proliferation of human iPS cells. Here the intuitive approach would be to use low agitation because iPS cells are very sensitive. Figure 5 shows some iPS cell numbers as a function of time. We tested four different agitation profiles. The best-performing profile did not have the lowest agitation. The solution is that you should test different agitations instead of assuming the lowest agitation will be best. At low agitation, there is a trade-off: low shear means low-mass transport off the gases and of nutrients. This underscores the importance of testing multiple agitation profiles.
The third example is how the stirring rate affects the proliferation of human iPS cells. Here the intuitive approach would be to use low agitation because iPS cells are very sensitive. Figure 5 shows some iPS cell numbers as a function of time. We tested four different agitation profiles. The best-performing profile did not have the lowest agitation. The solution is that you should test different agitations instead of assuming the lowest agitation will be best. At low agitation, there is a trade-off: low shear means low-mass transport off the gases and of nutrients. This underscores the importance of testing multiple agitation profiles.
In summary, cell therapy manufacturing can be quite complicated. But if you understand the biology and you test your conditions, success is definitely possible. Always think about the endgame: a commercial process that produces enough cells at a low cost of goods. Don’t assume anything, and tackle complexity by experimentation.
Maribel Rios is managing editor at BioProcess International; mrios@bioprocessintl.com.