 Manufacture of biopharmaceuticals using mammalian cells inherently incurs a risk of viral contamination during cell cultivation. If introduced, viruses can infect and replicate in cells used to produce a therapeutic protein or vaccine. The consequences of such contaminations can be dramatic. Not only can a company lose contaminated batches, but it also faces potentially extensive root-cause investigations, facility cleanup efforts, and introduction of preventive measures. Until contamination issues are resolved adequately, production should not be resumed, and facility downtime brings significant economic and capacity implications.
Manufacture of biopharmaceuticals using mammalian cells inherently incurs a risk of viral contamination during cell cultivation. If introduced, viruses can infect and replicate in cells used to produce a therapeutic protein or vaccine. The consequences of such contaminations can be dramatic. Not only can a company lose contaminated batches, but it also faces potentially extensive root-cause investigations, facility cleanup efforts, and introduction of preventive measures. Until contamination issues are resolved adequately, production should not be resumed, and facility downtime brings significant economic and capacity implications.
Unintended introduction of virus into mammalian cultivation processes must be prevented at almost any cost. To achieve that goal, it is important to understand the entire biomanufacturing chain. Only through scrutiny of each aspect can a company identify possible risks for introduction and then effectively mitigate those risks.
The need to ensure viral safety during production is well recognized by biomanufacturers and regulatory agencies around the world. A comprehensive guideline from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) defines a framework covering cell-substrate characterization, unprocessed bulk harvest testing, and viral clearance (1). Less well addressed in that document are elements such as segregation of processing steps and raw-material control requirements.
In two previous papers written by the BioPhorum Closed System team, we addressed segregation of manufacturing steps relating to the purification process (2, 3). Here, we examine more closely viral contamination risks related to what may be the most susceptible unit operation: the cell cultivation step in which viral amplification can occur.
The Back Door: Eukaryotic cells require complex media to grow and express target proteins. Cells used in biomanufacturing need an energy source, amino acids, micronutrients, vitamins, minerals, and sometimes growth factors. The complex mixtures of media used in mammalian cell culture usually cannot be sterilized thermally, which is a typical preparation step for microbial cultivation media.
Instead, cell culture media typically are sterilized by conventional sterilizing-grade filters with 0.22-µm or 0.1-µm pore sizes. Although such filtration methods are effective against microbes (bacteria and fungi), they are unable to remove all viruses fully if one or more raw materials are contaminated. That creates a “back-door” entry to viral contamination of cell cultures.
Even if animal-derived raw materials are eliminated from a process, there remains a risk of adventitious-agent contamination to many other raw materials used. Such contamination events can take place during manufacture of the raw materials themselves, in storage, and in their final use. Several operations comprise that final use: unpacking, weighing, dissolution, and transfer into manufacturing areas. Pest control, gowning procedures, and routine cleaning will reduce risk, but they do not represent absolute barriers to potential adventitious agents. Wherever humans are involved arises the risk of contaminating a product stream with viruses, bacteria, mycoplasma, and fungi.
Below, we present a case study in which a source of contamination was not identified during initial risk assessment of the biomanufacturing process. Once the risk was identified, mitigations were put in place immediately and effectively to prevent recurrence. Although we do present those mitigations herein, our primary aim is to highlight the necessity of full “360-degree” risk identification in biomanufacturing processes. Prevention is always better than cure.
Background and Problem Statement
A baseline guide on biopharmaceutical manufacturing facilities from the International Society for Pharmaceutical Engineering (ISPE) presents the modern concept of closed bioprocess technologies (4). Closed bioprocessing provides a higher level of process protection and a higher product safety profile than open processing can. By isolating a process and product from contaminants in the environment, closed systems also provide great opportunities to biopharmaceutical companies in efficiency, safety, and facility design. Closed bioprocessing allows for manufacturing in a controlled, nonclassified (CNC) environment because the surrounding environment no longer affects the process.
As defined in the ISPE guideline (4), a functionally closed system is one that may have been opened before product contact, but for which measures were taken to remove real or theoretical environmental contaminants that might have affected a process subsequently. A formal evaluation is required to confirm that the “closing” measures or operations are appropriate for mitigation of risks from the environment, personnel, and operations before product contact.
Media quality is critical to the success of cell culture and fermentation operations. Beyond their basic physicochemical properties, media must be free of bioburden, viruses, and other adventitious agents that could affect subsequent operations and quality of drug substances produced. Media preparation includes multiple steps that are typically performed in protected but open environments.
Media are collected from an open container (e.g., a drum, bag, or other container). This weigh–dispense operation typically is performed in an area that is separate from liquid-handling operations. Dispensing often is performed under high efficiency particulate air (HEPA) filtration, such as inside a dust-collecting downflow booth, in a Grade D environment. The process is protected further by operators dispensing media into single-use powder-handling bags while wearing personal protective equipment (PPE), including gloves that are changed between operations.
Dispensed media are introduced into a solutions preparation vessel that was cleaned in place appropriately before addition of process water and media powders. The latter may be added through an open port, a protected port (e.g., Powdertainer bioprocess containers from Thermo Fisher Scientific or containers from ChargePoint Technology), and/or a powder blending addition system (e.g., Tri-blender systems from TECNinox).
Media are blended in a protected vessel until dissolution is complete and pH is adjusted (taking minutes to hours), then sterile-filtered into a presterilized receiver (e.g., gamma-irradiated single-use or steamed-in-place multiuse vessels).
Some media also are subjected to thermal sterilization operations such as ultrahigh-temperature (UHT) (UHT), steam-in-place (SIP) and autoclaving, or thermal operations such as high- temperature short-time (HTST) pasteurization.
The risk of microbial proliferation in media preparation operations is low before hydration, so the duration between initial powder addition and completion of filter or thermal sterilization typically is limited and controlled. Expeditious “closing” of media (whether by filtration or thermal sterilization) ensures rapid removal of latent bioburden, thus preventing growth and ensuing detrimental effects such as media degradation and endotoxin formation.
In the case study described below, standard procedures were followed for media-preparation operations, yet subsequent cell culture operations were contaminated because of a failure to understand all risks inherent to the production process. It involved highly concentrated virus preparations, the infectious nature of which was well understood by the manufacturing company. Measures had been taken to ensure that virus-handling operations were well segregated (both spatially and operationally) from the “virus-free” host-cell culture/expansion areas. However, a lack of appropriate controls led to concentrated virus being introduced into the host-cell media-preparation area. Once they were introduced into the media, latent viruses could not be removed by sterile filtration, so subsequent host-cell cultures failed. That resulted in multiple batch-campaign failures.
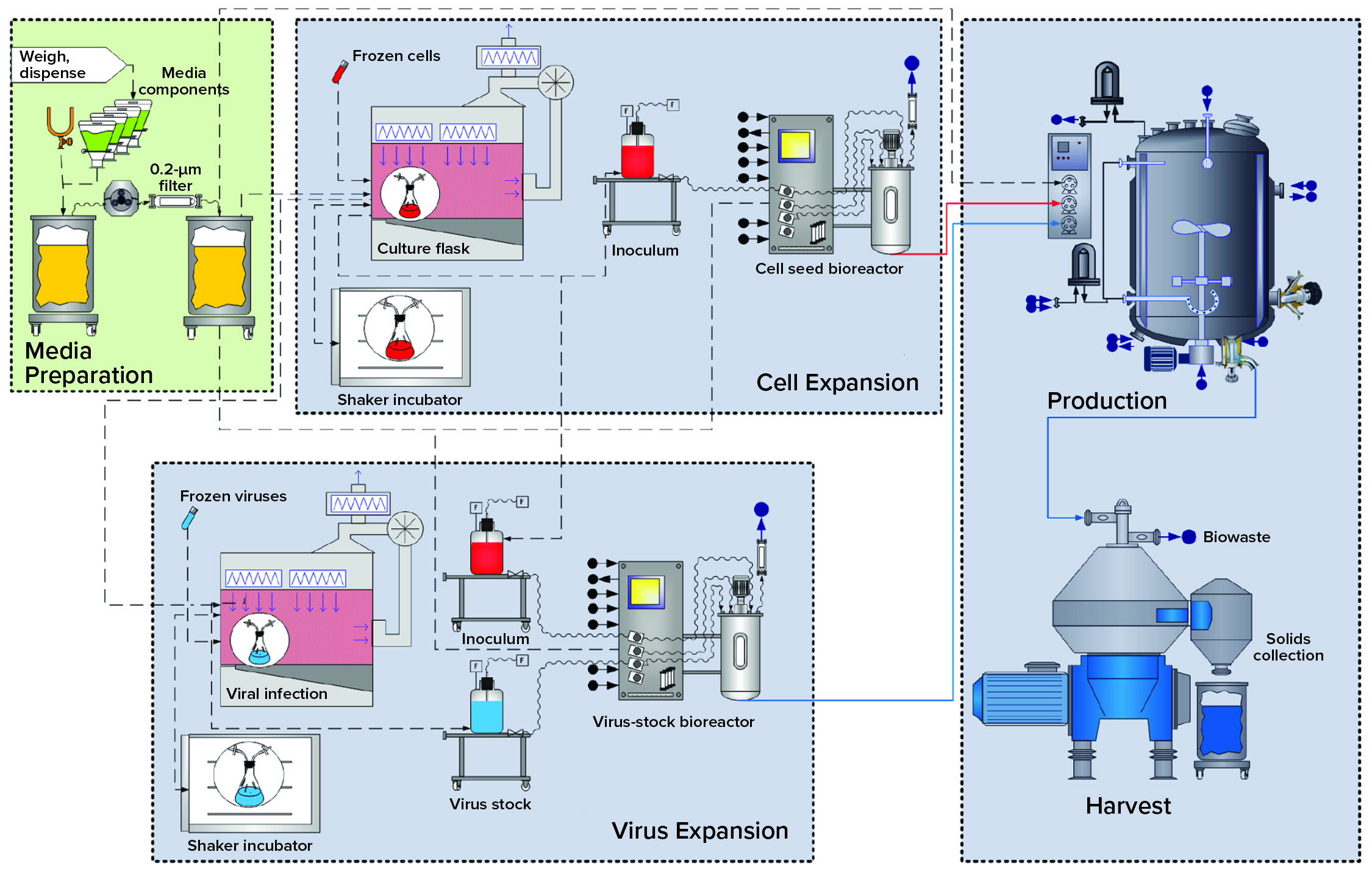
Figure 1: Overview of the baculovirus expression-vector system (BEVS) upstream process
Case Study — Spontaneous Infection
The baculovirus expression vector system (BEVS) is a commercial biomanufacturing platform by which a recombinant baculovirus is used to infect insect cells to induce expression of vaccines or therapeutic proteins (5). The technology takes advantage of a high-expression promoter of the baculovirus that can infect only insect cell hosts. The same Spodoptera frugiperda host insect cell line is used from product to product, making this an attractive system for producing commercial biopharmaceuticals.
BEVS Upstream Process: Figure 1 overviews upstream operations in the BEVS manufacturing process, which begins with media (and solution) preparation. Dry media components and water for injection (WFI) are dispensed and mixed in an open container. Formulated media are sterile filtered in an effective bioburden-reduction methodology that cannot remove virus particles. Latent virus particles in media solutions can pass through 0.22-µm filters.
Cell expansion occurs in shake flasks with open, aseptic sampling and transfer operations performed under a biosafety cabinet (BSC). Multiuse bioreactors used for cell expansion are cleaned and sterilized before addition of sterile media and before culture inoculation. All additions to and samplings performed outside the BSC go through aseptic connections.
Virus Expansion: Open aseptic virus-stock sampling and propagation operations are performed in a BSC. Bioreactors used for virus expansion are cleaned out of place and sterilized before inoculation and infection of host cells. All additions to and samplings from this bioreactor also go through aseptic connections.
Production: Final product is expressed by S. frugiperda cells in a bioreactor that has been rendered closed using clean-in-place (CIP) and SIP procedures. All additions to and sampling from the production bioreactor also go through aseptic connections.
Harvest: In this particular case study, the production-bioreactor contents are processed through a centrifuge, with cell slurry collected in an open container.
As Figure 1 shows, those host-cell expansion, virus expansion, and production and harvest operations are segregated by facility design (spatial separation) to mitigate the risk for unintended infection of the host-cell culture. Virus-containing manufacturing areas also are spatially segregated from other critical support areas, such as media preparation. That includes sterile filtration before inoculation.
In a BEVS process, a host insect-cell culture is expanded to required volume and density before baculovirus vectors are introduced to infect the host cells and initiate protein production. In this case study, an insect-cell culture of 500 L was sampled before that step to confirm that cell density and viability requirements were met before infection with the “Product B” baculovirus. Unfortunately, cell-viability, diameter, and morphology characteristics of the sample evaluated in this case study were indicative of insect cells that already had been infected with baculovirus. That preliminary finding was confirmed by subsequent genetic analysis, which identified the infection as “Product A” baculovirus used in the previous manufacturing campaign. So the company initiated an investigation to determine the root cause of that infection.
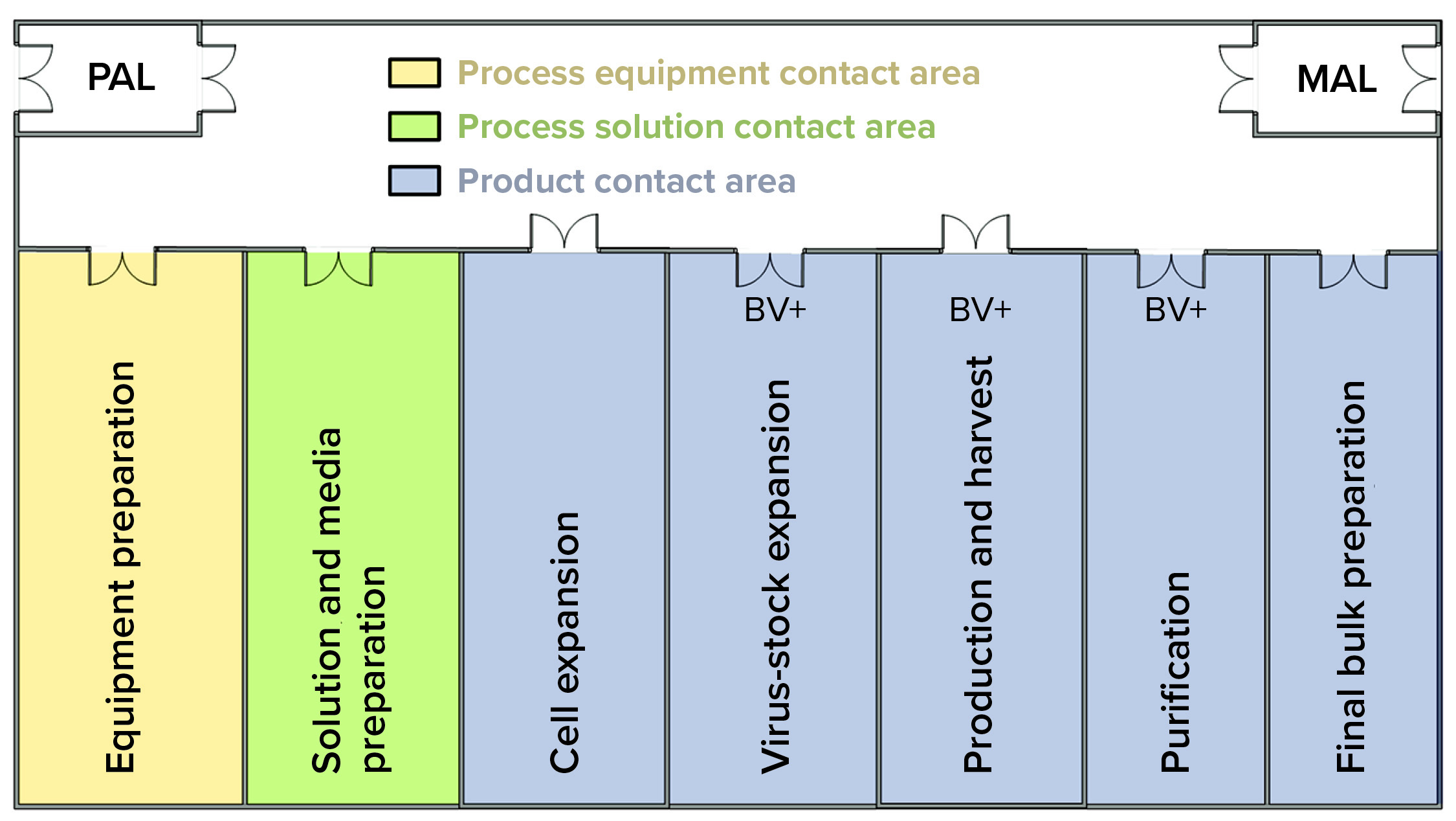
Figure 2: In this facility layout, operators enter through a single ingress/egress personal airlock (PAL) to enter the whole facility, where they don gowns, shoe covers, hair bonnets, and gloves before entering manufacturing suite. In the entry corridor, operators can travel between rooms as needed without passing through another PAL. If gloves or gowns appear to be soiled, then operators return to the PAL to refresh those as needed. Operators exit the facility using the same ingress/egress PAL. They use the material air lock (MAL) to bring raw materials, consumables, and equipment into the facility and to remove waste, samples, equipment, and final bulk drug substance.
The investigation included sampling and testing media batches used for host-cell maintenance and expansion. Results indicated the presence of contaminating recombinant Product A baculovirus in preinoculated, preinfected, sterile-filtered media. A batch-record review revealed that media used to culture the host insect-cell line at the Product B seed scale were prepared during harvest of the Product A production batch. Both activities could occur simultaneously because the work was performed in separate rooms of the facility (Figure 2).
Review of the batch records for the preparation of infected media and the Product A production batch harvest showed that both operations were performed on the same day by the same operator. Documented times in the batch records showed that those two operations did occur simultaneously. The operator was moving between the harvest room containing active Product A virus and the media preparation room (Figure 3).
The facility layout was intended to segregate media preparation from cell expansion — and from virus-active zones. Media and solution preparation activities were isolated from the virus-active zones for protein production and harvest. All areas were classified as Grade D consistent with the ISPE guideline for closed processing (4). The operator at the center of this investigation had over 10 years of experience with the company and was regarded as one of the most experienced and reliable operators in the production department.
The investigation turned to media-preparation operations. A combination of single-use and reusable systems was used in the open media preparations. The operation set-up involved installing a gamma-irradiated bag liner into an open 200-L plastic tank. An autoclaved mixer was introduced to the top of the lined portable tank, then media were hydrated. After pH and osmolality adjustments, media were filtered using a sterilizing-grade filter that had been tested to confirm integrity. Prepared media were stored at 2–8 °C until use (with a shelf life of eight weeks).
The harvest operation also was evaluated as a potential source of the contamination. The process harvest step is based on centrifugation with open collection of solids discharge from a centrifuge that contains the product. Solids collected consist of cells, cell debris, and live viruses. The overall duration of harvest is three to four hours, with intermediate storage of each discharge at 2–8 °C as a bioburden control measure. Those discharges cause some aerosolization and potential soiling of the immediate environment, including an operator’s outer garments and gloves. At this site, operators would spray their gloves periodically with 20% ethanol and wipe them with lint-free wipes to mitigate the risk of spreading virus to other areas. At the time, rules for personnel transitions between manufacturing areas included replacement of gowns and gloves that were soiled visibly. Note that the only glove- and gown-changing station was located at the bidirectional entry/exit airlock to the manufacturing area of the facility.

Figure 3: Operator movement caused a contamination event; (1) the operator performs open harvest operations from the product A bioreactor containing live virus;
(2) the operator migrates from harvest to media preparation areas without a gown-change transition; (3) the operator performs media preparation operations for the subsequent product B batch.
Investigation Outcome: The operator indicated that he had set up for media preparation after assisting with harvest of several cell-slurry discharges but that he had not replaced his outer garments in transition from the harvest area to the media-preparation area. Even extremely small volumes of virus can be hazardous because they can be amplified during cell cultivations. The likelihood of transferring microliter quantities of baculovirus-containing broth into media preparations was very high.
Sterile filtration cannot be an effective mitigator of that risk. The presence of baculovirus in culture media clearly resulted in an infection of host cells during cell maintenance or expansion. Even very low-level contamination would be amplified over several cell passages after cells had been maintained with the contaminated media — and further amplified when the insect cell culture was expanded. But the Product A virus did not reach a high-enough level to be detected until it reached the production bioreactor.
A lack of effective systems in place at the time was identified as the root cause for this cross-contamination event. Facility flows as shown in Figure 4 were found to present an unacceptable risk for viral cross-contamination. Gown and glove inspection monitoring were supposed to happen throughout the day but relied on operators’ interpretation of what is an “unacceptable” amount of soiling. The location of the gowning changing stations made it inconvenient to change gloves when transitioning between functional areas.
Setting up clear rules on transitioning between functional areas and providing airlock gowning-change stations at transition points reduces the likelihood of such contamination events. Making the right thing to do also the easy thing to do always improves compliance with standard operating procedures (SOPs).
Quantitative Evaluation of Carryover: Assuming that the root cause is carryover of in-process material from centrifuge operations into media preparations, we can quantify how much material it would take for carryover. Assuming a baculovirus concentration of 108/mL, then 1/108 of one milliliter would contain a single infectious virus. That corresponds to 10–9 L — or 1 nL. Such a low volume is but a fraction of a droplet (1 drop ≈ 20 µL), so the slightest invisible soiling of a glove that subsequently contacts a component going into cultivation media could suffice to elicit a contamination event.
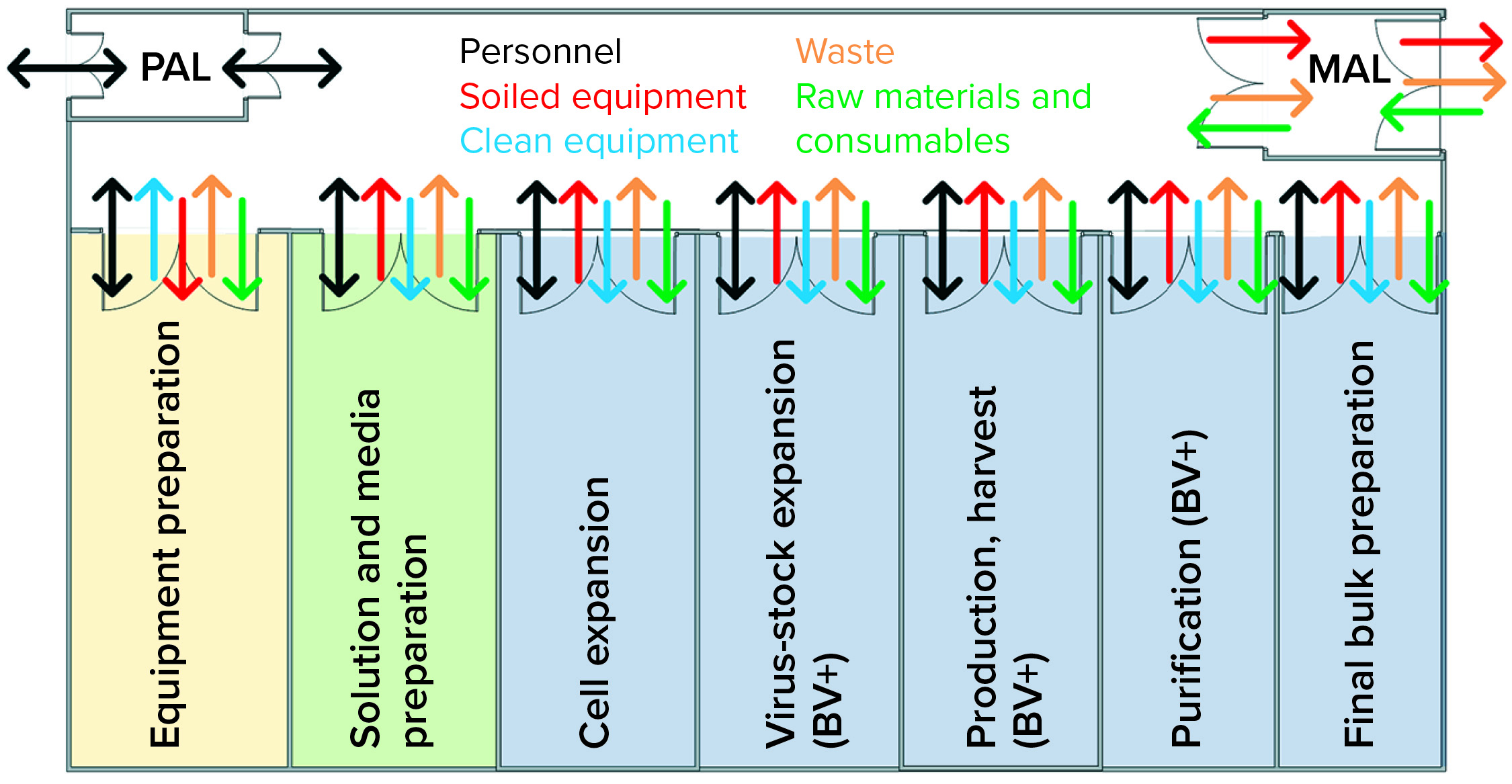
Figure 4: Case-study facility design does not promote effective transitions of personnel, equipment, or materials; barriers between baculovirus-containing areas and virus-free areas were inadequate, as depicted.
Technical Aspects
What type of risk-mitigation actions should be taken to remove latent virus-contamination risk in media preparation and bioreactor operations? The simple answer is to put more layers of protection into the process and facility. In this case, cell-cultivation medium is the most probable vector to carry viruses into cell-culture operations. This is not a unique risk. Other industries have used robust adventitious barriers such as HTST pasteurization, nanofiltration, and gamma/UV irradiation in media preparations.
HTST could be the most widely used technique for thermally inactivating viruses in media. During media transfer, heat (>80 °C) is applied to media for a specified duration (e.g., two minutes) for inactivation of viruses. However, HTST is efficient only in high-volume applications and requires high capital expenditure to install and validate the necessary technology.
Nanofiltration is another option to remove undesirable viruses from media. Special filters have been developed for use with cultivation media, combining high throughput and low cost. Nanofilters retain viruses while media pass through. Based on the media components and their absorption to nanofilters, the effects of this process on filterability and cell-culture performance could differ from case to case. Therefore, compatibility studies need to be performed to assess the applicability of nanofiltration technology in a given process.
Irradiation can be used to inactivate potential adventitious contaminants in media components. It is used frequently for serum, in fact. Irradiation produces electron disruptions (ionization) that kill organisms by damaging their DNA and other cellular structures. This method is not used widely, however, because it also may cause degradation of components within a medium.
Multiple layers of protection should be considered in areas where cross-contamination can occur through human interventions and reuse of equipment. For example, if a previous batch has been contaminated with undesirable viruses, and if associated equipment (e.g., tanks and small items) are not cleaned well enough, that contamination could transfer to the next batch. Therefore, multiuse equipment and small items should be cleaned with a virus-inactivating solution and then autoclaved or steamed in place wherever the risk of virus carryover is possible. If equipment cleaning is determined to be a root cause, then single-use systems might be considered a mitigation — but they are not a panacea.
In the case study above, the cross-contamination still would have occurred if the process had been in single-use systems exclusively. Closing (to protect a process from its environment) and containing (protecting the environment from a process) high-risk process steps such as centrifugation harvest is a highly effective mitigation strategy. Instead of collecting cell-slurry discharges in an open vessel, a closed collection strategy could be engineered to reduce the potential for operator exposure to contaminating harvest materials, thereby reducing the probability of cross-contamination.
Moreover, appropriate gowning would reduce the risk of contamination from human intervention. Such procedures should be in place, and operators should be trained to prevent possible introduction of adventitious agents. If some activities must be separated in a facility, using different colored gloves and gowns can be effective visual cues to prevent noncompliance. It becomes obvious that SOPs are not being followed when an operator is wearing the wrong color of gown or glove. Such errors can be corrected immediately by anyone observing the discrepancy.
Wherever facility constraints make complete physical segregation of activities impossible, temporal procedures can be implemented. In our case study, the excursion might have been prevented by a simple rule: “Media preparation cannot be completed simultaneously with harvest.” Before starting media preparation, daily cleaning must be performed and harvest activities completed in a prevalidated amount of time. Timing such activities before initiating media preparation operations would ensure that sufficient air changes have occurred to clear the room air of potential airborne contaminants. The goal of facility layout and procedures for reducing contamination risk should be to make it easy for operators to do so in practice. A facility designed such that it is difficult or impossible for operators to go to an area without breaking rules is an example of poor engineering controls. Figure 5 presents a facility layout with improved controls.
In addition to engineering controls, several procedural controls can be implemented to further mitigate the likelihood of cross-contamination (e.g., the different-colored gowning mentioned above). If a facility is designed to force operators to make gowning transitions, then the probability of an occurrence such as that described herein is mitigated.

Figure 5: This improved facility layout is designed to mitigate the risk of viral cross-contamination. Here the risk from baculovirus (BV) is mitigated by segregating BV activities from those related to cell expansion, solution and media preparation, and final bulk preparation. Materials moving from BV+ to BV-free areas first would need to be either cleaned and sanitized in the equipment preparation area or have their exterior surfaces sanitized (using isopropyl alcohol spray/wipes) and wiped down in the material airlock (MAL) between BV-free and BV+ areas. BV-free intermediates moving from purification to final-bulk preparation would need to go through the same sanitization procedure when they transition from the BV+ to the BV-free area. Documents summarizing those sanitization procedures and flows would need to be approved and validated to ensure that segregation can be maintained.
Little Things Mean a Lot
Even in carefully controlled environments, sometimes things can go terribly wrong. In the clear light of hindsight, we might wonder why the contamination event in the case study above was not foreseen. Simple mitigations could have been put in place that — with high probability — could have prevented transfer of virus to the cultivation media preparation operation. Once the procedural changes and facility changes were implemented, in fact, operations have continued for over 10 years in that plant without another such “spontaneous infection” event. Those changes necessitated hiring of additional operators to provide for proper segregation between viral-positive and viral-negative activities.
It is common practice for media preparation to be performed as a relatively open operation and followed immediately by sterile filtration. That step ensures that microbial contamination will be removed rapidly, preventing growth and the ensuing detrimental effects on culture media such as degradation or endotoxin formation. Standard procedures are followed for media preparation operations, including gowning and the use of gloves. Sterilizing-grade filtration of the complete growth medium was applied as is usual for cell-growth media. These measures were insufficient, however, to mitigate the risk of viral contamination in our case study.
Manufacturing processes involving highly concentrated virus preparations are not typical. In more conventional manufacturing operations, proteins of interest are secreted from recombinant cell lines. No viruses are required because the gene construct for producing the therapeutic protein is contained in the cell line itself. Even if live recombinant cells were transferred from a production area to media preparations (highly unlikely because eukaryotic cells usually do not survive such trips), such cells would be removed effectively by filtration of the final cultivation medium.
We believe that the root cause of the failure above was a combination of insufficient “routine manufacturing practices” and the important contribution of a highly concentrated virus in downstream materials. Those risks were not identified properly for evaluation during assessment of the entire process. Recognizing such a failure mode requires virological insight and an understanding of the quantitative aspect of transfer discussed above. So it is not surprising that the risk was overlooked. A number of other risk factors had been identified and mitigated properly — but it took only a small oversight (e.g., on the order of magnitude of a nanoliter) to miss this failure mode.
Learning from mistakes is always painful; learning from the mistakes of others is much less so but underlines the value of sharing this type of information. By discussing such events publicly, our industry as a whole can benefit from hard-earned experiences of individual companies.
References
1 ICH Q5A(R1). Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. US Fed. Reg. 63(185) 1998: 51074; https://database.ich.org/sites/default/files/Q5A%28R1%29%20Guideline_0.pdf.
2 Boulanger R, et al. Shared Clean-in-Place Systems: To Share or Not to Share? BioProcess Int. 18(4) 2020: 16–22; https://lne.e92.mwp.accessdomain.com/manufacturing/facility-design-engineering/shared-clean-in-placesystems-to-share-or-not-to-share.
3 Kamstrup S, et al. How Much Harm Can a Single Droplet Do? Considerations for a Viral Inactivation Step. BioProcess Int. 18(10) 2020: 46–52; https://lne.e92.mwp.accessdomain.com/downstream-processing/viral-clearance/how-much-harm-can-a-single-droplet-do-considerations-for-a-viral-inactivation-step.
4 Baseline Guides. Volume 6: Biopharmaceutical Manufacturing Facilities — Second Edition. International Society for Pharmaceutical Engineering: North Bethesda, MD, November 2013.
5 Chambers AC, et al. Overview of the Baculovirus Expression System. Curr. Protoc. Protein Sci. 21 February 2018; https://doi.org/10.1002/cpps.47.
Lead Authors: Rob Boulanger is a process specialist, and Marc Pelletier is director of CRB. Søren Kamstrup is a principal scientist at Novo Nordisk. Buyoung Lee is a principal engineer in the manufacturing science and technology (MSAT) network at Genentech (a member of the Roche group).
Contributors: This paper was written as a consensus view of the BioPhorum Closed System team. The main contributors included the following people. Alejandro Kaiser is a CRB process engineer. Benjamin Avery is associate director and senior manufacturing manager of Partner Therapeutics. Corresponding author Tim Corbidge is a BioPhorum facilitator (tim.corbidge@biophorum.com). Christopher Gilliard is a bioprocess technician III at AGC Biologics. Robert Hendrix is a senior design engineer for single-use technologies in the bioproduction group at Thermo Fisher. Per Kærsgaard is a research scientist at Novo Nordisk. And Eric Raaijmakers is a process-support lead in upstream processing at Merck.
The case study described in this paper originates from a company other than those with whom the authors are affiliated currently, and it has been adapted for publication.