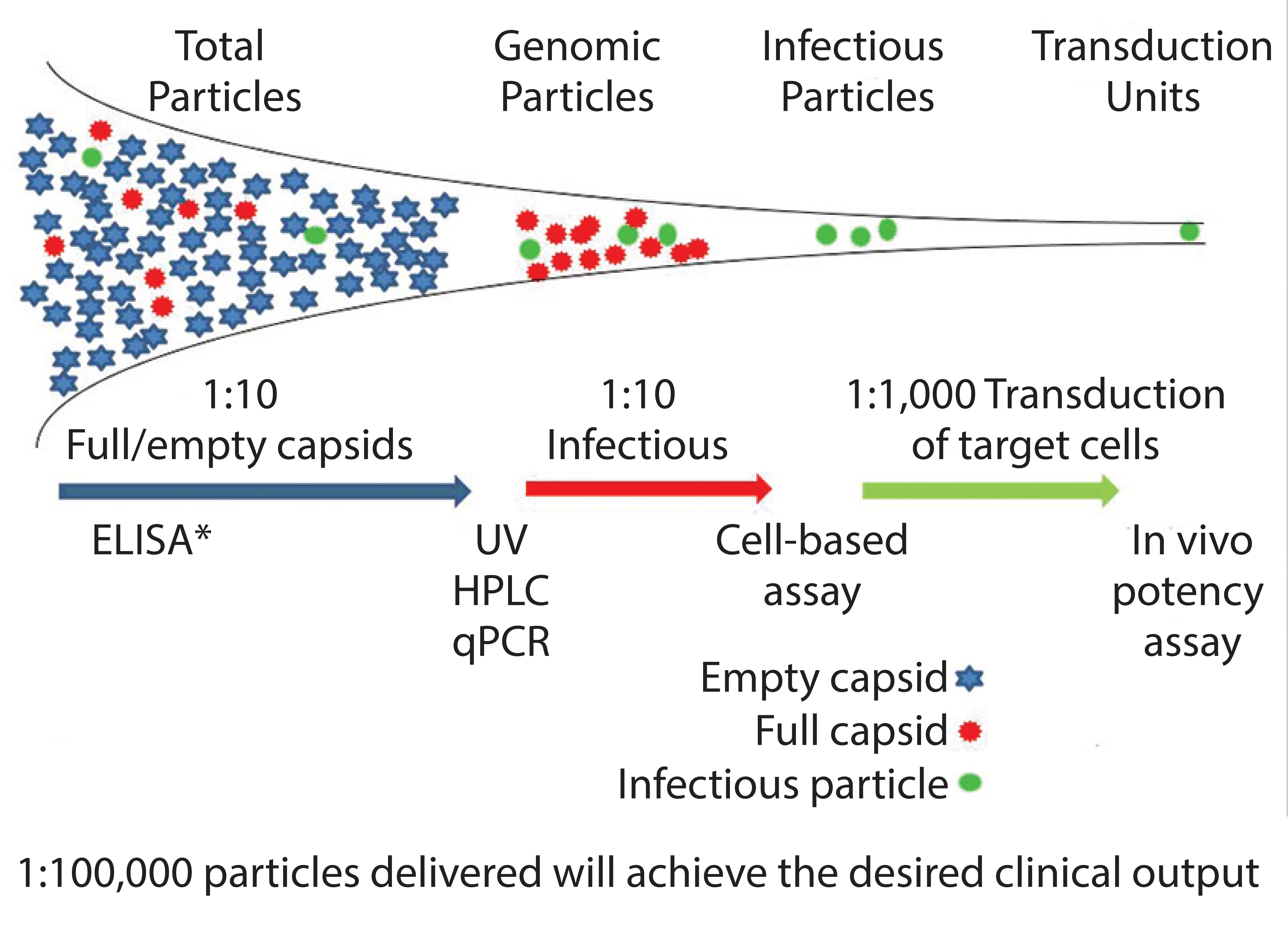
Figure 1: Vector heterogeneity * ELISA determines total particles. qPCR determines empty particles.
After many years of development, gene therapy is beginning to deliver on its promises in the clinic, in some cases with spectacular outputs. Those clinical successes also have led to an influx of funding and engagement from large pharmaceutical companies, thereby bringing the required financial support and expertise for late-stage clinical developments and product commercialization. Although many initial studies were confined to small patient groups and focused on a range of rare monogenetic diseases, new approaches to gene editing have significantly broadened potential use of adenoassociated virus (AAV) mediated gene therapy to a broader range of therapeutic applications, specifically in immunotherapy.
Consequently, process development groups and manufacturers now are faced with the transition of such products into licensed clinical products that are available, safe, and affordable to broader patients groups worldwide. Manufacturing approaches for AAV products have been based on adherent cell culture processes that often are combined with centrifugation recovery and purification procedures. Such strategies have provided a simple and relatively low-cost approach to meet vector supply needs for small proof-of-principle clinical studies, often involving <10 patients. However, those approaches are not suitable for large-scale production of clinical grade AAV, especially for diseases such as haemophilia and muscular, liposomal storage disease — for which dose levels can be as high as 1015 vg/patient.
The shortfalls in manufacturing capabilities for viral vectors are well documented (1), and significant investment is targeting the development of new production platforms, which are hoped to will generate scalable and cost-effective platforms for future clinical products. The immediate issue for a number of product developers is the transition from their current production approaches to supplying material for early phase clinical studies to ones that can be used for commercial supply. That is not just a case of increasing process scale, but also of addressing quality expectations for marketed biotherapeutics. Existing guidelines relating to the manufacture of gene therapy products identify the need for increased characterization of products (including product-related impurities) and the potential for integration of noncoding sequences into vectors (2).
Product heterogeneity occurs within all biologic manufacturing processes. Regulatory agencies require manufacturers to identify the source of heterogeneity and minimize levels through improved process understanding and process control. The majority of those product-related impurities appear to arise from failures in cellular process within virus production processes rather than during downstream processing (DSP) operations. Although DSP operations usually can reduce host contaminants with conventional purification approaches, removal of product-related contaminants often is significantly more challenging and not always successful.
Viral vectors (including AAVs) are unusual as therapeutic entities. Because of their high levels of product impurities, only a small proportion of the vectors produced can bring about the desired clinical benefit. And only a small proportion of vectors produced from the manufacturing process can infect the target cells and achieve expression of the desired gene product (Figure 1).
Heterogeneity is seen with AAV vectors at a physical level in terms of full, empty, and incomplete capsids and at a genetic level through the presence of undesired or incomplete sequences being packaged into the vectors, including antibiotic resistance genes from the plasmid vectors and host cell DNA (3). Such heterogeneity is well documented and may be acceptable within research environments, but it would be less acceptable for a licensed product — both in terms of overall levels of contaminants and batch-to-batch variability.
Although process development studies will continue to strive toward achieving higher productivity levels, there is clearly an increased need to identify routes to control and, if required, improve product quality. For some products, such requirements might take precedence over increases in process scale. With a product as complex as a viral vector, trying to identify, quantify, and understand root causes of heterogeneity might not be feasible, especially at a microscale level. What we will initially need is to focus on are key contaminants that are believed to impact patient safety and potency.

Table 1: Viral vector production routes
AAV vector production can be achieved through multiple approaches. Although the majority of vector products currently are made using transient transfection routes, other approaches to use include baculovirus, helper virus, and producer cell lines.
Irrelevant to the specific route of vector production, the actual processes for virus assembly within a cell are something we understand at only a relatively high level (4). This is especially true for key factors that affect assembly and packaging efficiency, including points of failure in the pathways involved in capsid construction and synthesis of the viral genome. Consequently, product developers have to take empirical rather than theoretical approaches to identify factors affecting vector heterogeneity and the operational ranges for key process steps to control it.

Figure 2: Production stages in transient AAV manufacturing
Vector Design
The initial factor that will affect heterogeneity of a vector is its design in terms of the capsid proteins and genetic payload it carries. Vector tropism is a key attraction of AAVs. A broad range of serotypes and increasing hybrid and engineered serotypes is used to improve targeting and to maximize expression of the desired protein within the target tissue (5).
Serotype differences affect productivity and point of vector accumulation — whether intracellular or secreted into the media. What seems to be less well documented is the effect of capsid engineering on the heterogeneity of vectors produced, including efficiency of capsid protein assemblies, the packaging of genomes, and stability of vectors once assembled. That effect may be critical, and the concept of “manufacturability” assessment (commonly applied for protein therapeutics including antibodies) might need to be performed as part of the vector-selection process for synthetic vectors.
An other element is the genetic content of the vectors, especially for vectors generated through multiple plasmid transfections. A number of studies have highlighted the potential for incorporation of antibiotic resistance genes and noncoding sequences into vectors (5). Despite existing regulatory guidance identifying that antibiotic genes should not be used if possible, the reality is that most viral vectors currently in development that are made using plasmid transfection still do contain antibiotic resistance genes.
Transgene size also can be a key factor with regard to levels of empty and full capsids, specifically where large therapeutic constructs are being applied, including those that exceed the natural limit of 4.8 kb. Such “oversized” vectors can decrease packaging efficiencies and result in increased levels of empty capsids being generated and incomplete genome packaging (3).
For vectors made through transfection routes, one approach to reduce misincorporation of noncoding sequences has been the use of minimized plasmid backbones without antibiotic genes, use of semisynthetic approaches such as minicircle DNA (7, 8), and synthetic approaches such as the Doggybone plasmids from Touchlight Genetics (9). In theory, these approaches can be introduced into on-going clinical development programs without compromising vector design or gene payload. They also can improve processes with smaller plasmid sizes. Such novel approaches are likely to become increasingly attractive. They may be cost effective in the long run with process optimization and in-process control, but they also may take some time to achieve.
Table 2: Comparison of cotransfection or serial transfection processes
Virus Production
The virus production phase of the manufacturing process for vectors produced through plasmid transfection can be divided into six key stages: cell build, transfection, virus production, harvest, purification, and formulation. The first four elements can be the most critical for controlling virus heterogeneity.
Essentially, developers can take one of two approaches to address product heterogeneity in biological processes. The first approach is to reduce the levels generated within the initial manufacturing process. The other is to remove product-related impurities in downstream processing. For viral vectors, many current production strategies rely on the latter option, using approaches such as density gradient ultracentrifugation to reduce levels of empty capsids. Although that option can achieve significant reduction in the level of empty capsids at laboratory scale, studies have not reported whether comparable levels of purification can be achieved with commercially available large-scale continuous centrifuges. The alternative and more conventional approach to removing product-related impurities is by using a chromatography system. However, although analytical and laboratory procedures for separating empty and full capsids can be conducted, achieving required levels of resolution at production scale is challenging. So the logical starting point must be to reduce the level of heterogeneity ahead of DSP operations while recognizing the limitations of those operations.

Figure 3: Upstream production stage and process variables for transient AAV vector production
From both processing and regulatory perspectives, manipulation of cell culture processes is an attractive approach. It can enable manufacturers to develop and enhance their processes without introducing “significant” process changes and allows for process improvements to be introduced postregistration. The complexity of cell culture processes provides significant scope for potential process improvement (Figure 3).
Processes can be improved not only by reducing impurity levels, but also by increasing consistency in those levels. Process consistency is a critical issue with products of this type because it relates directly to product potency and patient safety. High levels of product-related impurities can lead to failed batches, disrupted product supply chains, and high replacement costs. Looking at the initial phases of a production process (cell build and plasmid transfection), it is necessary to maintain cells in an optimal state and maximize their growth rate ahead of plasmid transfection. The most obvious route to increasing productivity is to increase cell density. As with other cell culture processes, that can be achieved through development of media supplements that prevent key metabolites from limiting cell growth and vector production.
We also need to understand optimal growth conditions for the transfection of plasmids. It is believed that the cell cycle stage can be critical to plasmid transfection, with the need to transfect when cells are at their most susceptible (possibly between the S and G2 phases). Thus, by synchronizing cell culture, we could increase virus productivity and quality. We also need to look at the transfection process itself.
Plasmid transfection also should be optimized, including the ratios and amounts of plasmids used in a process. Key factors include the amount of each plasmid (Helper, RepCap, and transgene) used to transfect cells and the ratios in which they are used. Most protocols simply identify the quantity of plasmid used in a transfection process, and in some protocols that is titered against cell numbers. However, that does not take into account variations in the sizes of plasmids used. Therapeutic plasmid sizes will vary from around 4.8 kb to 6.8 kb — which is a 40% range in vector size. It seems logical to base plasmid level on copy number per cell rather than on total weight to ensure that plasmid transfection ratios can be retained. AAV production through triple plasmid transfection requires establishment of viral replication and capsid formation capability within a selected producer cell line through the Rep/Cap plasmid and helper plasmids as well as generation of a viral transgene containing the desired transgene.
In wild-type systems, AAV is produced when a cell has a preexisting AAV genome integrated into its chromosome and is replicated when the cell becomes infected with adenovirus or herpes simplex virus, with helper functions from the helper virus enabling AAV replication to take place. Because those are not simultaneous events, why should we perform plasmid transfection to achieve viral vector production as a singular event? Initial studies using serial transfection approaches at Cobra Biologics have shown that significantly reduced levels of empty capsids can be achieved through transfecting the transgene plasmid 24 h after the Rep/Cap and Helper plasmids.
Can we identify cellular media and nutrient requirements through metabolite and metabolomics studies to retain cell health during the virus production phase? Studies have shown that plasmid transfection does not overwhelm the cellular mechanisms in the way that viral infection does. Therefore, we can maximize the virus production phase and consequently increase viral productivity without compromising quality of vectors produced.
Such studies indicate that it is possible to increase product quality through systematic assessment of cell culture and the virus production process to identify critical process parameters. However, the opportunities for process improvement still depend heavily on the availability of suitable assays to determine vector titers and for vector characterization. The precision, reproducibility, robustness, and throughput of viral vector assays still remain a major hurdle for the industry and are arguably a higher development priority than cell culture or downstream development. Without access to meaningful assays, not only upstream development but also optimization of downstream operations will be infeasible.
From a process development focus, key assays correlate to the measurement of full and empty capsid levels. Although such assays may not indicate actual product potency, in theory they should act as indicators of the infectivity fitness of the vectors to deliver genes as desired. The challenge is not only to address single assay issues, but also to establish profiles of ratios of full to empty capsids and the proportion of full capsids as well as levels of misincorporation of DNA sequences that can infect target cells and achieve the desired gene expression. That knowledge then can be used to support development programs that can be targeted to improve ratio or to retain them when process changes are implemented.
In the near future, ongoing investments in process development will yield significant improvements in viral vector manufacturing platform scales, titers, and production costs. In the interim, we need to address issues of vector quality and those of increased scale and titers. In doing so, we can gain a much greater insight into the critical process parameters affecting our processes. Although undoubtedly challenging, pragmatic and defendable approaches can be used to achieve those goals for products already in clinical development — for both vector designs and manufacturing processes that do not overly compromise ongoing clinical programs.
References
1 Van der Loo JCM, Wright JF. Progress and Challenges in Viral Vector Manufacturing. Hum. Mol. Genet. 25(R1) 2016: R42–52; doi:10.1093/hmg/ddv451.
2 EMA/CAT/80183/2014: Guideline on the Quality, Nonclinical and Clinical Aspects of Gene Therapy Medicinal Products (draft). European Medicines Agency: London, UK, 23 March 2015.
3 Schnödt M, Büning H. Improving the Quality of Adeno-Associated Viral Vectors Preparations: The Challenge of Product-Related Impurities. Hum. Gene Ther. Methods. 28(3) 2017: 101–108; doi: 10.1089/hgtb.2016.188.
4 Bennett A, et al. Understanding Capsid Assembly and Genome Packaging for Adeno-Associated Viruses. Future Virology online 28 June 2017; www.futuremedicine.com/doi/pdf/10.2217/fvl-2017-0011.
5 Kotterman M, Schaffer DV. Engineering AdenoAssociated Viruses for Clinical Gene Therapy. Nature Rev. Genet. 15, 2014: 445–451.
6 AAV Vector Production: State of the Art Developments and Remaining Challenges. Cell Gene Therapy Insights 2(5) 2016: 521–551.
7 Schnödt M, et al. DNA Minicircle Technology Improves Purity of Adeno-Associated Viral Vector Preparations. Mol. Ther. Nucleic Acids 5(8) 2016: e355.
8 Munye MM, et al. Minicircle DNA provides Enhanced and Prolonged Transgene Expression Following Airway Gene Transfer. Sci. Rep. 15 Mar 2016: 23125; doi: 10.1038/srep23125.
9 Lukashchuk V, et al. Next-Generation DNA Constructs for the Rapid and Safe Manufacture of AAV Vectors for Regenerative Gene Therapy. British Society for Cell and Gene Therapy (BSCGT) conference, Cardiff, Wales. April 2017.
Corresponding author Tony Hitchcock is technical director at Cobra Biologics, Stephenson Building Keele Science Park, Keele ST5 5SP, UK; 44-1782-877288; www.cobrabio.com. Vera Lukashchuk and Kevin Bowes are principal scientists, research and development at Cobra Biologics.