On the brink of bringing exciting new therapies to commercialization, cell therapy developers are taking notice of how other companies are addressing processing and technical challenges. Here, leaders from Dendreon, Advanced BioHealing, and Pluristem describe their current cell therapy programs. And two organizations —the Alliance for Regenerative Medicine (ARM) and McLaughlin–Rotman Center for Global Health —provide details on the promises of regenerative medicine.
Cellular Immunotherapy
Dendreon’s Provenge (sipuleucel-T) cell therapy induces an immune response to aid in treating existing prostate cancers. The US Food and Drug Administration (FDA) classifies it as an autologous cellular immunotherapy, although the company also uses the term active immunotherapy to distinguish it from a preventative immunotherapy such as a vaccine. Dave Urdal, chief scientific officer at Dendreon (www.Dendreon.com), explains its processing.
BPI: How does time to manufacture become a critical factor?
DU: Key raw materials that go into a dose of sipuleucel-T therapy come from a patient’s blood. The final product is live cells, never frozen, so its shelf life is much shorter than that of a recombinant protein. Antigenloaded activated cells must be infused into a patient within 18 hours from the time we complete cell harvest.
BPI: What is involved in the processing?
DU: First, we isolate white blood cells from a patient’s blood using apheresis. The white blood cell fraction is collected and sent to a Dendreon manufacturing facility. At our facility, we remove residual red blood cells and granulocytes from the preparation using buoyant density centrifugation. We isolate antigenpresenting cells (APCs) and culture them in a recombinant antigen (Figure 1). That cell fraction is placed in culture with defined media made of a genetically engineered recombinant protein composed of the prostate antigen linked to a cytokine known as granulocyte-macrophage colony-stimulating factor (PAP-GM-CSF). Those cells with that antigen are then cultured for about 40 hours. During culture, the GM-CSF portion activates the APCs within that mixture of cells. Those cells take up antigen and load up the surface of the APCs with peptide epitopes that “educate” the T cell compartment of the immune system
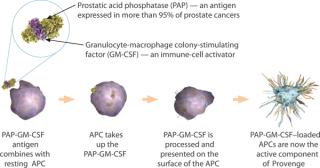
BPI: How do you ensure cell quality?
DU: Key operating steps include maintaining cells in the right type of growth media and at the right conditions to ensure they remain viable and have ideal characteristics. We use procedures similar to those of stem cell transplantation, in which hemapoeitic stem cells are isolated from bone marrow or peripheral blood. We manipulate cells in culture using centrifugation. Those are common steps used in conventional biologics manufacturing, but our work is done under conditions that minimize cell damage.
We’ve worked out assays to help us verify the process was taking place for each dose. We have a very specific potency assay to show that cells are activated for each patient. Each dose meets prespecified quality release criteria before the lot is released for infusion into the patient. We use a straightforward approach of assessing cell viability in preparations, and we enumerate the cells. So we monitor a number of those types of properties on every batch before release.
BPI: What is next for Dendreon?
DM: The success has validated our approach of using autologous APCs in ways that will be useful for treating other cancers. We have filed an investigational new drug application (IND) for another antigen for targeting HER-2 positive tumors. The next clinical testing is in patients with invasive bladder cancer. We have other target antigens that are in preclinical stages of development that would allow us to address treating colon cancer, renal cell carcinoma, and other malignancies.
Dermal Healing
Relaunched in 2007, Advanced BioHealing’s (ABH’s) Dermagraft therapy is a dermal substitute used to treat diabetic foot ulcers. Kathy McGee, senior vice president of operations at ABH, provides an overview of its processing and safety testing.
BPI: What is Dermagraft?
KM: Dermagraft is a cryopreserved human fibroblast-derived dermal substitute; it is comprised of fibroblasts, extracellular matrix, and a bioabsorbable scaffold. The final product is an approximately 2-inch × 3-inch piece of dermal tissue, approved by the US Food and Drug Administration for the treatment of diabetic foot ulcers.
BPI: What is the source of ABH cells?
KM: The fibroblast cell line used in the manufacture of Dermagraft was derived from a single newborn foreskin tissue, which was qualified in 1993 when the product was owned by Advanced Tissue Sciences. From the donor tissue we isolated the fibroblast cells—the cells that make up the dermal layer of skin —to create a master cell bank and then expanded the cells to generate a working cell bank. The same cell line has been used in the manufacture of Dermagraft since the product was initially launched in 2001, and is fully qualified under the Center for Biologics Evaluation and Research’s Points to Consider.
BPI: What are the processing stages?
KM: The Dermagraft manufacturing process begins when fibroblast cells are removed from the working cell bank. Our daily routine includes thawing cells from the working cell bank and expanding them further to grow enough cells to seed onto our final product. We begin with a starting point of about 1 million cells and over a period of about four weeks, expand and grow the cells in roller bottles to result in approximately 1 billion cells to begin producing our final product.
The cell expansion process is in a monolayer. As we start our growth process, we harvest the cells and feed them onto a three-dimensional bioabsorbable mesh scaffold that will break down in the body when implanted into the patient’s wound. When the cells are introduced onto the scaffold, we maintain the environment to closely resemble as much as possible the conditions they would see in the human body. We maintain the pH and temperature and add the nutrients and various concentrations of nonessential amino acids and growth factors that the cells need to form a healthy dermal tissue For 2.5–3 weeks, we allow the cells to grow in a closed system, which becomes part of the product’s final configuration. Part of the final packaging is placed into a foil pouch and then into a cardboard box, labeled, and frozen to –70 °C. The tissue is frozen using a cryo-preservative, which helps protect the cells during freezing and thawing
BPI: What testing is done to ensure the final product is safe and efficacious?
KM: Before releasing the product, we extensively test for efficacy and safety per predetermined and approved specifications. There is a therapeutic range and safety profile that the product must meet before its release to ensure the product has not been compromised during processing. Our safety profile ensures our product is safe for a host
of patients; we have no history of patients experiencing rejection issues. Further, there is no pigmentation, so it doesn’t matter which patient gets the product.
BPI: How is sterility ensured?
KM: Our cells and all of our starting materials are considered sterile, and all of our processing components are disposable. We use preassembled, irratiated bioreactor pouches to which we aseptically add the cells. The product is grown in the pouch, which also serves as its final packaging configuration (Photo 1).
Photo 1:
Allogeneic adult Stem Cells for “Personalized” Therapy
Pluristem Therapeutics (www.pluristem.com) uses adult stromal cells derived from donor placenta to produce cellular “drug factories.” Although the company does not yet have a product on the market, the potential for its technology to treat a variety of diseases and indications is drawing interest. Zami Aberman, CEO, describes how the cells are processed and discusses plans for further clinical trials.
BPI: How can Pluristem’s adult stem cell therapies be used in treatment?
ZA: Our cells act as tiny (20-μm cell size) drug manufacturers. We are using adult stem cells as sophisticated drug delivery vehicles. When cells are injected into a patient’s body, they receive biomechanical signals from the patient and start to produce a specified drug as the patient needs it. The problem of over- or under-dosing is prevented because delivery is based on a patient’s individual biomechanical signals that direct the quantity needed. So the treatment becomes a “personalized therapy.”
BPI: How is processing conducted?
ZA: We start with a donor’s consent. We take blood samples from a mother to ensure she does not carry biocontaminants. After the placenta arrives at our facility, we keep it for three weeks. During that time we check for potential contamination. We extract cells, clean them, and grow them in two-dimensional Petri dishes to rid them of all immunological ingredients. Only then do we culture them in our unique bioreactor technology, harvest them, and cyropreserve them. They are kept for one year, and during that time they are sent to a hospital or other facility, where they are thawed and injected.
Our process produces high quantities of cells with batch-to-batch consistency. Because the final product does not require a match, we can produce it at one location and ship it around the world. From one placenta we can treat about 10,000 patients for a variety of indications.
BPI: What therapeutic areas are you targeting, and what is the progress in those areas?
ZA: We’ve completed phases 1 and 2, proving safety and defining the dose for the next study, which will start during the first half of 2011. The first clinical study is treating patients with critical limb ischemeia (CLI), which is related to peripheral arterial disease (PAD). CLI patients have pain or nonhealed ulcers on their feet.
We have followed both FDA and European Medicines Agency (EMA) guidelines. We conducted phase 1 and 2 studies under FDA and under EMA approval. Phase 1 was conducted using 1-L bioreactors, but now we are approaching 5-L bioreactors, and at marketing we will use 15–20 L bioreactors.
BRAZIL’S REGENERATIVE MEDICINE INDUSTRY
McLaughlin–Rotman Center for Global Health (in association with the University of Toronto) recently conducted a study on regenerative medicine (RM) in Brazil (1). Dominique McMahon (researcher, PhD student) conducted face-to-face interviews with 50 RM experts, including researchers, policy analysts, funders, companies, and regulators for that study.
BPI: What are the main therapeutic areas of Brazil’s regenerative medicine industry?
DM: One main driver of RM in Brazil has been the need for new technologies and new products to address local health challenges. There has been a shift of Brazil’s health profile toward chronic disease because of economic growth that led to changes in diet and lifestyle, and due to decreasing burdens of infectious and parasitic disease. In the 1930s, ~50% of all deaths were related to infectious and parasitic disease; in 2005 it dropped to 5%. Cardiovascular disease has increased threefold since the 1930s and is currently the cause of about 32% of all mortality.
Clinical research continues in cardiology, orthopedics, diabetes, and neurology. In addition, one treatment in clinical trials is for Chagas disease, which is a local tropical disease that can cause serious heart problems if left untreated. Brazil has conducted several clinical trials using stem cell treatments and had one of the largest stem cell trials in the world for cardiomyopathies. Brazil has also developed a treatment for diabetes mellitus which when tested in a small group of patients, allowed long-term insulin independence, and a promising stem cell treatment for stroke.
BPI: What are the stengths in Brazil’s program?
DM: Our study found that Brazil is doing quite well in RM work, but it is an emerging sector. Some of the economic, regulatory, and policy events have been very supportive. For example, the growing strength of the economy has made possible investments in biotech (which are considered higher risk and which require significant infrastructure). Recent laws that support human embryonic stem cell research, and the innovation law which helps develop linkages between universities and industry are important. But the field has moved forward because of clear government support and the need to develop solutions to local health problems.
BPI: What are areas of improvement?
DM: If this sector is to do well, it must have consistent financing. It is very difficult to find private funding in Brazil; grants are almost entirely from public sources. Researchers are often on very short-term grants, which forces them to define their research agendas with short-term goals.Other major barriers to developing RM products in Brazil include the cost and delays of material importation. Researchers claim it can sometimes take up to a year for them to receive items they have ordered. More collaboration between firms, researchers, and clinicians may also help the development of RM therapies.
BPI: How would you describe Brazil’s regulatory environment?
DM: Brazil has a strong regulatory environment. The equivalent to US FDA is ANVISA (National Health Surveillance Agency), the main drug regulatory group. Stem cell therapies are not considered drugs, but ANVISA does oversee all of the laboratory facilities that generate cells for clinical use. The Ministry of Health oversees clinical trials and is responsible for approving RM therapies. The other main regulatory body is CONEP (Coordinator, National Ethical Committee), which oversees ethics. All stem cell clinical trials must gain national ethics approval from the CONEP board.
Reference
1 McMahon D, et al. Regenerative Medicine in Brazil: Small but Innovative. Regen. Med. 5(6) 2010: 863–876.
Regenerative Medic
ine
The Alliance for Regenerative Medicine (ARM) was established in 2010 as a group of companies and other institutions that aimed to draw attention to the progress being made in regenerative medicine (RM), new stem cell therapies, and tissue engineering. According to ARM chairman Gil van Bokkelen, the group also provides industry with a voice, particularly in Washington, DC.
BPI: What are ARM’s objectives?
GVB: We’re focused on policies that relate to legislation introduced at the end of the last congress that was designed to promote some aspects that are important to ARM — specifically, achieving legislative clarity around the Dickey–Wicker amendment. We are also promoting funding and trying to create incentives for investment into the RM area. On the regulatory side, we are working with the FDA so that we can establish clearer standards for companies that are developing new therapies. Investors don’t like uncertainty, so having clear regulatory standards that enable companies to operate efficiently and cost effectively is very important. Companies need to do that while bearing in mind the primary goal, which is to protect patient safety. So we think working directly with the FDA and learning from them but also working to educate them in terms in exciting new technologies.
BPI: Is ARM working toward educating the general public about the possibilities of regenerative medicine?
GVB: That is another important objective. ARM is involved in educating the media and the general public so that they can understand these new technologies. I think a lot of people intuitively understand the potential for regenerative medicine and what new therapies might mean in healthcare. I also think there is a very high degree of comfort among people in the public about the potential for stem cell medicine and regenerative medicine technology.
One of the facts we’re trying to convey through multiple different forums and other efforts is that regenerative medicine really has transformation potential in the sense that it’s one of the few areas where we can both dramatically improve the clinical outcomes in high-need areas while simultaneously shifting the cost curve in the right direction.
BPI: Is ARM addressing questions about the safety of RM products?
GVB: Many of these technologies are associated with an outstanding safety profile. If you look at the history of drug development, the odds of developing a small-molecule therapeutic historically have been about 1 in 20 or 1 in 10 at best. But in many instances, you are developing small molecules that don’t normally exist in the body — they are synthetic constructs. However, if you look at the success rate of biologics that haven developed during the past couple of decades, the success rate is actually substantially higher than that, so the odds of developing a biologic have been more than 20% — that is from the time you start clinical development. One reason for that is that many of these therapeutics have a good safety profile — they are substances that normally exist in the human body. That’s the differentiating factor. With respect to stem cells, many therapies being developed are based on what ordinarily exists in the body. We are just harnessing the power of specific cell populations, turning them into a “pharmaceutical-grade product,” and applying them in a way that makes sense to speed recovery and healing. So the emerging safety profile is something that a lot of people view as being very exciting. And now what they are looking for is evidence of efficacy or therapeutic effectiveness in a range of areas that make the case in a compelling way. But that evidence is starting to emerge across various studies.
REGENERATIVE MEDICINE: ADDRESSING UNMET MEDICAL NEEDS
Gil Van Bokkelen, director of the Alliance for Regenerative Medicine, describes an example of how regenerative medicine can help in tissue healing.
“When most people think about RM, they tend to think first about inserting a new organ or inserting new tissue to replace something that has been lost. Although, there are certainly some exciting possibilities in that regard, the application of RM is a lot broader than that. Certain cell types can home to sites of tissue damage, inflammation, and injury. In many instances these therapies could be administered locally, injecting them near or right into the site of damage, or administered intravenously, where the cells will respond to various signals in the body and go where they are needed. The cells express multiple types of proteins or factors that can promote or accelerate healing and tissue repair — shifting a body’s healing response in the right direction, for example by reducing inflammatory damage, and promoting formation of new blood vessels.
“RM has the potential to address a wide range of unmet medical needs. For example, one area that illustrates the potential of these new therapies would be in the area of treating patients that have suffered an ischemic stroke. In the United States every year there are ~800,000 people that suffer a stroke, of which ~85% are caused by a clot or a blockage in an artery (~2 million people if you include Europe and Japan). Unfortunately, if you suffer an ischemic stroke, you only have about a three-hour window to get to a doctor to get treated with the only drug that is currently available to a stroke victim (tPA, Genentech). If you can’t get to the doctor in that three-hour time frame, you are not supposed to receive treatment with tPA because by that point it could actually do more harm than good. So as a practical consequence, ~95% of the people that have an ischemic stroke are not treated with tPA because they can’t get to a doctor in time. Essentially these patients receive palliative care, which ranges from physical therapy to extended hospitalization or even permanent institutionalization.
“The average cost of care for a stroke victim is in the hundreds of thousands of dollars. For those that can’t afford or don’t have access to long-term institutional care, a family member will have to care for them, and in many instances people live with a dramatically reduced quality of life. It’s a huge cost for our healthcare system — about $73 billion a year just for stroke, according to American Heart Association. And the numbers are expected to skyrocket in the years ahead given the aging population.
“Several companies are focusing on adult-derived stem cells that can be administered to stroke victims. Preclinical research from studies has shown that in some instances, stem cell therapy not only reduces or corrects a lot of the damage from stroke, but it can lead to a nearly complete recovery. Moreover, evidence suggests some stem cell therapies can be administered several days or even a week (or possibly longer) after a stroke has occurred and generate those results. Imagine what that might mean for those who have suffered from a stroke. That could change the landscape of stroke medicine as we know it.”
Author Details
Maribel Rios is managing editor of BioProcess International, 646-957-8884; mrios@bioprocessintl.com.