 Virus removal/inactivation is a major concern in the safety of monoclonal antibodies (MAbs) and other recombinant-protein drugs. Some methods (such as nanofiltration and low-pH inactivation) have been demonstrated repeatedly by the industry to be reliable for most viruses, with >4 log10 removal. Based on my company’s virus-removal experiences with its MAb downstream-process platform, we propose a “bracketing method” — testing only samples that lie at the extremes of a design space — to prove proactively that small differences in operating parameters among different product manufacturing processes do not affect the virus log10 reduction value (LRV). At our company, we determine LRV based on our experience and data from our virus removal/inactivation platform to ensure the viral safety of biopharmaceuticals in development.
Virus removal/inactivation is a major concern in the safety of monoclonal antibodies (MAbs) and other recombinant-protein drugs. Some methods (such as nanofiltration and low-pH inactivation) have been demonstrated repeatedly by the industry to be reliable for most viruses, with >4 log10 removal. Based on my company’s virus-removal experiences with its MAb downstream-process platform, we propose a “bracketing method” — testing only samples that lie at the extremes of a design space — to prove proactively that small differences in operating parameters among different product manufacturing processes do not affect the virus log10 reduction value (LRV). At our company, we determine LRV based on our experience and data from our virus removal/inactivation platform to ensure the viral safety of biopharmaceuticals in development.
At the investigational new drug (IND) stage of development, virus-safety studies are performed to demonstrate acceptable levels of safety for clinical trial materials (CTMs). Virus detection and clearance verification should be carried out in accordance with ICH Q5A principles; however, in the early stage of research, no proof of robustness is required (1, 2). Therefore, pharmaceutical companies can use a platform process strategy to evaluate viral safety. When developing biotherapeutics, companies can ensure viral safety of their manufacturing processes and marginally shorten development times with prior knowledge.
We established a preclinical viral clearance platform process based on our biomanufacturing process development experience with the purification of more than 10 MAbs using the same host cell line and similar serum-free culture media. Capability verification data for low-pH virus inactivation, nanofiltration, and downstream column chromatography fully demonstrate that MAbs manufactured by this platform process have good viral safety and controllability for clinical research use.
In the clinical research stage of a new coronavirus-neutralizing antibody “MAb-X,” we use a bracketing method to demonstrate viral safety of the manufacturing process. Based on platform data, our company intends to accelerate process development, move into clinical research with a plan for continuous process improvement during this stage, and conduct comprehensive virus-removal process verification.
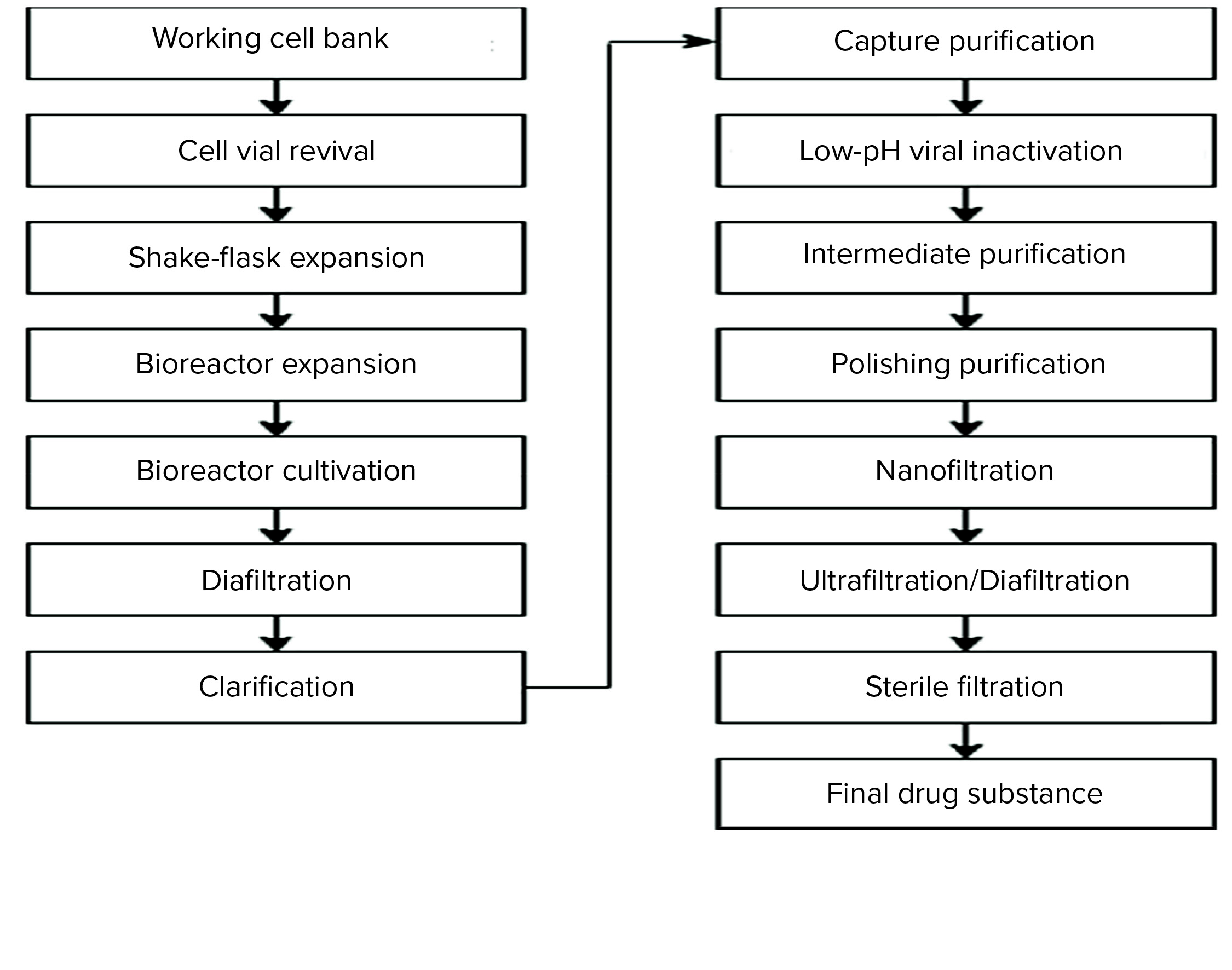
Figure 1: MAb-X drug substance production process flow chart
MAb-X Production and Target Quality Overview
MAb-X is expressed by high-density suspension culture of Chinese hamster ovary (CHO) cells in a fed-batch process. The product is extracted and purified by clarification of cell-culture supernatant and a three-step purification process based on protein A affinity and ion-exchange chromatography. The downstream process includes two orthogonal virus removal/inactivation methods — low-pH inactivation and nanofiltration — and concludes with ultrafiltration to obtain bulk drug substance. Filling and stoppering of the drug product are performed in an aseptic facility. The drug is formulated to be administered intravenously, so its dosage form is a solution for injection of 10–15 mg/kg of recipient body weight. MAb-X antibody product-related impurities are controlled with reference to general indicators:
- monomer purity of ≥95%, as measured by size-exclusion high-performance liquid chromatography (SEC-HPLC) and sodium-dodecyl sulfate capillary electrophoresis (CE-SDS)
- acidic charge isomers ≤30%.
Process-related impurities are controlled as follows:
- host-cell protein (HCP) residual ≤100 ppm
- host-cell DNA residual ≤10 ng/dose
- protein A shedding residual ≤100 ppm.
In addition, we apply the ICH Q5A “three-tier viral-safety control” principle (2, 3): documenting virus-introduction points, process-control technologies, and clearance verification. The MAb-X drug-substance production process-control strategy includes
- cell bank virus verification (in accordance with ICH Q5A guidelines for endogenous and exogenous virus detection)
- viral safety considerations for production culture medium (no serum and no animal-sourced materials, independent postvirus media-formulating area)
- virus titer detection and infectivity tests in cell culture harvest (external source virus central control detection)
- production plant equipment control (current good manufacturing practices (CGMPs) cleanroom, workshop, and single-use closed production equipment)
- personnel (well-trained operators).
In addition to low-pH virus inactivation and nanofiltration, the affinity, anion-exchange, and cation-exchange chromatographic steps have proven capacity for removal of different types of viruses. Figure 1 is a process flow chart for the MAb-X drug substance to be commercialized.
Control of Virus Introduction Risk in Production Process: From potential virus introduction through process control to the removal/inactivation process during MAb manufacture, a complete virus-safety risk-minimization strategy can be formed (4). Using our minimization strategy based on the ICH Q5A implementation guidance, we analyzed our platform technology and evaluated the effectiveness of the viral safety control strategy in the clinical research stage of MAb-X.

Cell Bank: The CHO cell line is used widely in development and production of many engineered antibodies and recombinant proteins. This expression system has the characteristics of correct assembly, folding, and glycosylation of antibodies expressed by mammalian cells. The cells can be grown under serum-free conditions with high levels of protein secretion, making them favorable for large-scale production. Most MAb drugs on the market are expressed and produced by recombinant CHO cell lines.
Safety testing of our MAbX CHO cell line found no contamination of bacteria, fungi, mycobacteria, mycoplasma, or relevant viruses. In addition, isoenzyme identification of lactate dehydrogenase (LDH), glucose-6-phosphate dehydrogenase (G6PD), and nucleoside phosphorylase (NP) detected no cross-contamination. Table 1 summarizes the test results.
Our company has developed and established both the master and working cell banks independently. The cell line was constructed according to our platform-based CHO cell-line construction technology. No adventitious or endogenous viruses have been found in either the cell banks or end-of-production cells (EoPC) during testing of the more than 10 MAbs that we have developed so far. Those results comply with the relevant ICH guidances and conclusions generally demonstrated by the biopharmaceutical industry and different regulatory agencies. We will test our cell banks according to ICH guidelines for virus detection during the phase 1 clinical trial.
Culture Medium: The serum-free medium used for MAb-X cell culture is a platform medium. All materials (basic and supplementary) are controlled internally (through materials inspection and supplier selection/audit). The medium is composed primarily of amino acids and inorganic salts, with no animal ingredients, thus eliminating related adventitious virus risk.
Virus Detection in Cell Culture Supernatant: In the past, we engaged the services of well-known good laboratory practice (GLP) laboratories both in China and abroad to test for endogenous retroviruses and adventitious viruses in our 200-L, 500‑L, and 3,500-L bioreactors. Test results were negative for adventitious viruses, indicating that the cell line was not infected. Retrovirus detection by transmission electron microscopy (TEM) showed that unprocessed supernatant produced by the platform process contained retrovirus and virus-like particles at titers ≤7 log10, which was in line with general detection levels of CHO cell lines. During the clinical phase, we will implement adventitious virus testing for all clinical lots according to the ICH guidance.
Facility, Equipment, and Personnel: MAb-X clinical drug substance and drug product both are produced in a CGMP facility. Equipment, facilities, and clean areas have been qualified, and personnel have been fully trained. The drug-substance production line uses fully closed and disposable production equipment. The vial-filling line’s dilution and filtration systems also are disposable. Control measures based on risk assessment are followed to prevent the introduction of viruses, microorganisms, and other exogenous contaminants.
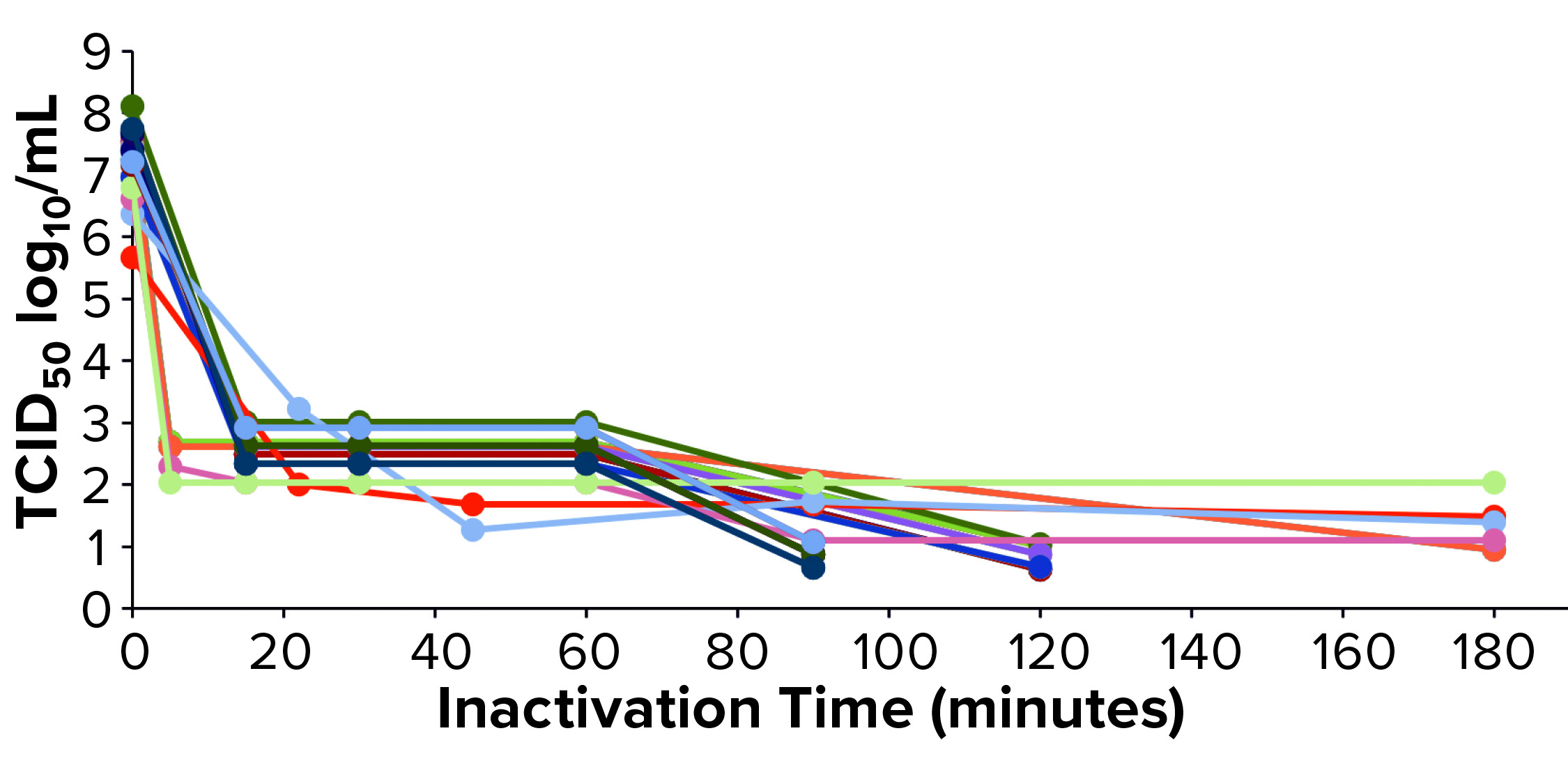
Figure 2: Xenotropic murine leukemia virus–related virus (X-MuLV) low-pH inactivation kinetic curve for multiple MAbs
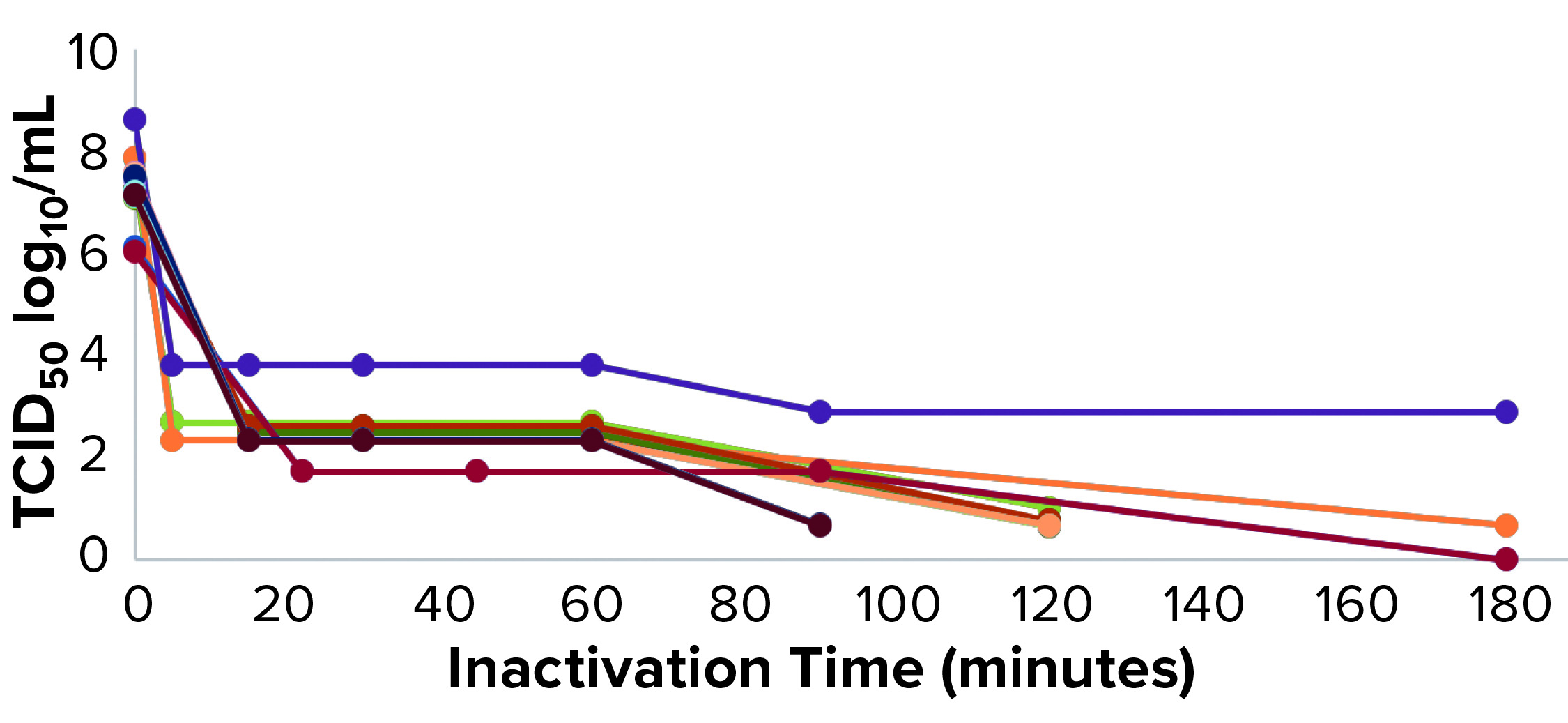
Figure 3: Pseudorabies virus (PRV) low pH inactivation kinetic curve for multiple MAbs
 Virus-Removal Evaluation of Manufacturing Platform
Virus-Removal Evaluation of Manufacturing Platform
Our company has completed viral-clearance studies for at least two orthogonal steps of its platform for more than 10 MAb products in early developmental and has completed full viral-clearance evaluation for three products in late-stage development.
Low-pH Virus Inactivation Process Platform Performance: For establishing a low-pH virus-inactivation process, the influence of pH on protein quality needs to be considered to ensure that it will remain relatively stable during the incubation process. At the same time, we must ensure that set incubation conditions can inactivate viruses to meet requirements.
In a typical MAb downstream process, a protein A affinity column is used for product-capture chromatography, with generally low-pH elution (pH 3–4, protein A harvest solution pH 4–5). Therefore, a low-pH virus-inactivation process (pH 3–4) comes after the protein A affinity chromatography harvest to reduce the necessary pH adjustment of the harvest solution.
For the low-pH virus-inactivation process, we set the minimum pH range and longest processing time according to protein stability data. For comprehensive virus inactivation, it generally is recommended that the pH is ≤3.8, incubation time is ≥30 minutes, and the incubation temperature is ≥14 °C. The buffer system should use acetate or citrate, the total protein concentration should be ≤40 mg/mL, and the NaCl concentration should be ≤500 mM (5).
We have used protein A purification and low-pH virus inactivation to conduct a virus-inactivation study on 13 MAbs at the IND stage — of which three have had complete process verification before biologics license applications (BLAs) were filed. We used two lipid-enveloped model viruses — pseudorabies virus (PRV) and xenotropic murine leukemia virus–related virus (X-MuLV) — as recommended by ICH Q5A (2). Figures 2 and 3 chart the batch information for verification of the low-pH inactivation process of those 13 MAbs.
We used a fixed 0.02 M citrate buffer for protein A affinity chromatography elution. Judging from multiple batches of affinity chromatography harvest solutions, the highest protein concentrations were different among the MAbs tested, ranging from 8.46 g/L to 23.62 g/L. That indicates the range of binding strengths for different molecules and protein A ligands, thus resulting in different elution harvest volumes.
In the current study, protein concentration at the time of inactivation was related to stability of the target protein and not to the effect of virus inactivation (6). MAb products usually are somewhat tolerant to low-pH buffer systems because of their relatively high isoelectric points. Results from our historical products also show that the target protein shows no change in its quality attributes after an inactivation time >180 minutes (data not shown).
Judging from the inactivation effect of a lipid-enveloped indicator virus, the pH conditions for viral inactivation generally are selected from a range of 3.5–3.7, and the inactivation temperature is 18–25 °C (“room temperature,” although laboratory definitions of that can differ). For inactivating viruses, pH 3.7 and 18 °C can be regarded as the worst-case conditions in this platform; pH 3.5 and 25 °C can be regarded as the optimal conditions for inactivation.
| Viral Inactivation |
The inactivation process takes place after MabSelect SuRe LX protein A chromatography (Cytiva) elution and harvest. Critical parameters are controlled as follows:
Other parameters are controlled as follows:
|
Figures 2 and 3 show the low-pH inactivation results of X-MuLV and PRV for 13 platform process MAbs as a function of time. Different pH values in the range of 3.5–3.7 at different laboratories showed no significant differences. When the inactivation time reaches 60 minutes, the inactivation titers of both X-MuLV and PRV have reached to >4.3 log10 for 10 MAbs. With longer inactivation times, different MAbs show increasing inactivation values to different degrees. In all cases, virus-reduction levels of ~4 logs are obtained as early as 5–10 minutes. A clear trend of log reduction can be seen from 60 to 90 to 120 minutes for most studies. Figure 2 shows the reduction curve of X-MuLV from all 33 runs (including a duplicated run for MAb 5, lot 3); Figure 3 shows the 32 PRV runs. The “Viral Inactivation” box describes our results from this study.
Statistical Analysis: Using IBM SPSS statistics software (version 23) and a multivariate analysis model, we analyzed the factors that affect LRVs of X-MuLV: e.g., batches, time points, antibody concentration, and pH. The model considers time point, antibody concentration, and pH as covariates and uses time point as a repetitive effect.
Tables 2 and 3 show the statistical analysis results, the former representing those obtained within first 60 minutes, and the latter representing those obtained within 180 minutes. No factor affects LRV significantly within that first hour; only inactivation time has a significant effect within two hours.
Those results show that different MAbs, inactivated pH levels, and protein concentrations within the scope of this study do not affect virus inactivation significantly, although inactivation time is a significant factor.
Based on our multiproduct LRV results and statistical analysis, we use our MAb platform process to evaluate the effect of the lipid-enveloped virus inactivation process for clinical sample production of MAb-X. The proposed process parameters are listed in the “Virus Inactivation” box.


Nanofiltration Process Platform Data: Viral membrane filtration technology contributes to biological product virus removal and inactivation (7). Currently the most reliable virus removal technology, it exploits the difference in particle size between viruses and proteins to pass through the latter (which are smaller than the average pore size of a membrane) while trapping the former (larger than the average pore size) to remove viruses. Membranes with an average pore size of ~20 nm serve as parvovirus filters. With increasing demand for removing small viruses, they have been implemented widely in the biopharmaceutical industry.
The virus-removal efficiency of nanofiltration is affected by parameters such as transmembrane pressure, protein concentration, ionic strength (conductivity) and pH of a solution, and operating time. Transmembrane pressure (TMP) is a critical parameter in the process. According to the Parenteral Drug Association, high transmembrane pressure is detrimental for virus filtration (8). Therefore, “constant-pressure filtration” is the common mode for nanofiltration virus removal to ensure that membrane pressures will not rise along with filtration amounts.
The effects of protein concentration are different across projects, but if it is too high, it will compromise the filtration operation. Other parameters such as feed pH, conductivity, filtration temperature, and time are not critical. The key acceptance standard for nanofiltration virus removal is the filtration volume per unit area (in L/m2/h, LMH). The physical interception method of nanofiltration is depends on the membrane material: Different membranes can handle different filtration volumes that can be blocked or even penetrated by viruses.
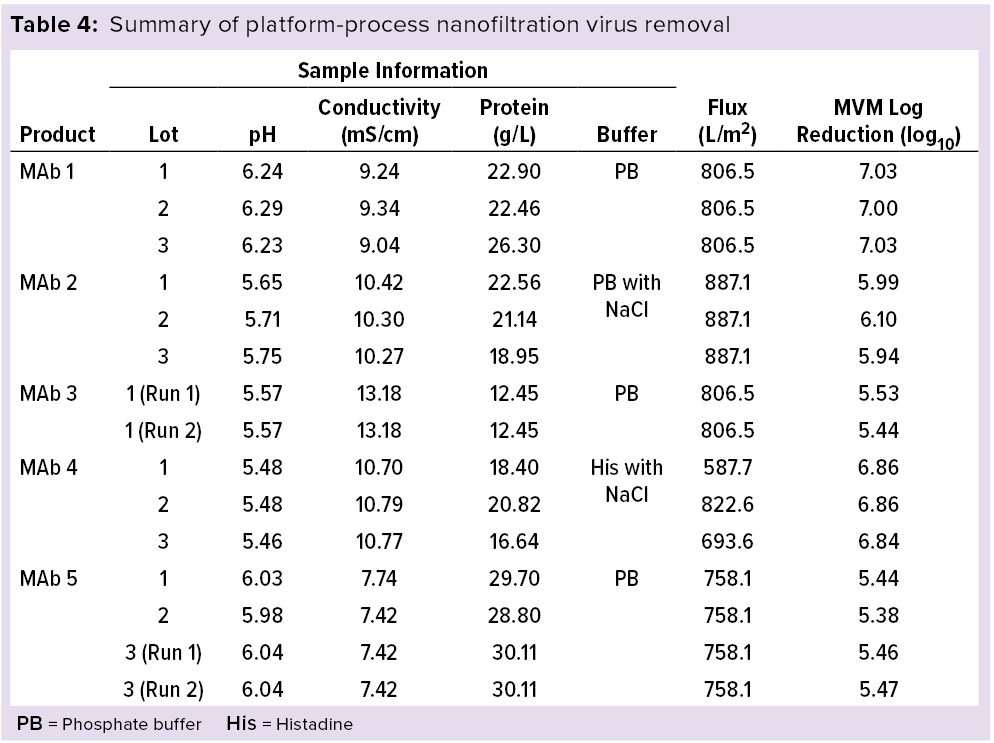
My company’s MAb manufacturing platform process model includes an IND-stage evaluation of virus nanofiltration effects. Selection of an indicator virus is based on membrane pore-size retention and ICH Q5A guideline recommendations for two viruses: minute virus of mice (MVM, 18–24 nm in diameter) and reovirus type 3 (Reo-3, ~70 nm). Table 4 summaries our research results.
In the development of multiple MAbs, we tested and verified the removal effects of virus-removal membrane filters from multiple manufacturers: Viresolve Pro nanofiltration membranes from Merck/MilliporeSigma, SV4 nanofiltration membranes from Pall, HF nanofilters from Sartorius Stedim Biotech, and Planova BioX filters from Asahi Kasei Bioprocess. Based on the multiple representative data available, for this study we used a Viresolve Pro membrane for MAb-X virus-filtration assessment.
| Viral Filtration |
The nanofiltration process follows cation-exchange chromatography and uses a phosphate/histidine buffer system either with or without sodium chloride. A well-known, widely applied filter is used. Critical process parameters are controlled as follows:
Other parameters are controlled as follows:
|
Because of its physical retention principle, nanofiltration assessment depends on the purity of a spiking virus used. That will be different for indicator viruses cultivated in different commissioned laboratories. For our verification study of IND-stage virus filtration using MVM and Viresolve Pro nanofiltration membranes, we selected the WuXi AppTec and BioReliance laboratories. Table 4 compares data from five MAbs (15 runs).
The Viresolve Pro nanofiltration membrane process uses a constant pressure of 40 psi. As mentioned above, the other critical parameters are membrane material and flux, which are closely interdependent. We used a platform technology that controls the effectiveness of virus removal. For the 15 runs of five MAbs in Table 4, we used cation-exchange chromatography as a purification step before nanofiltration. The different elution conditions had no significant effect on nanofiltration flux within the scope of the study. While maintaining 40-psi constant pressure, the platform process uses a flux ≤580 L/m2 to ensure reduction of >5.3 log of MVM.
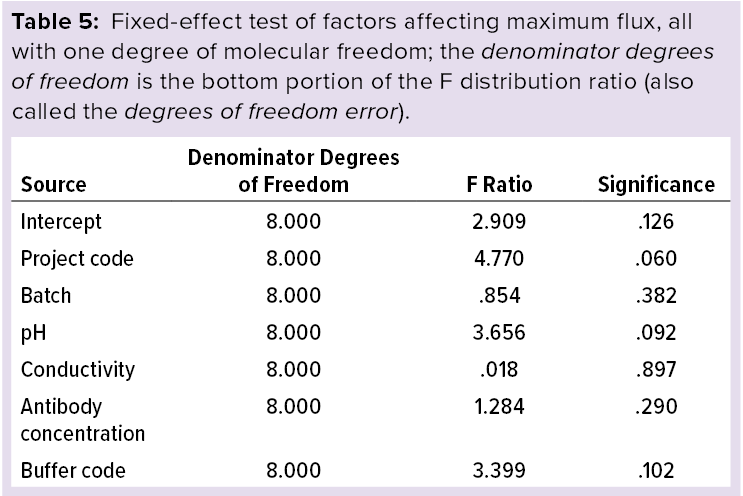
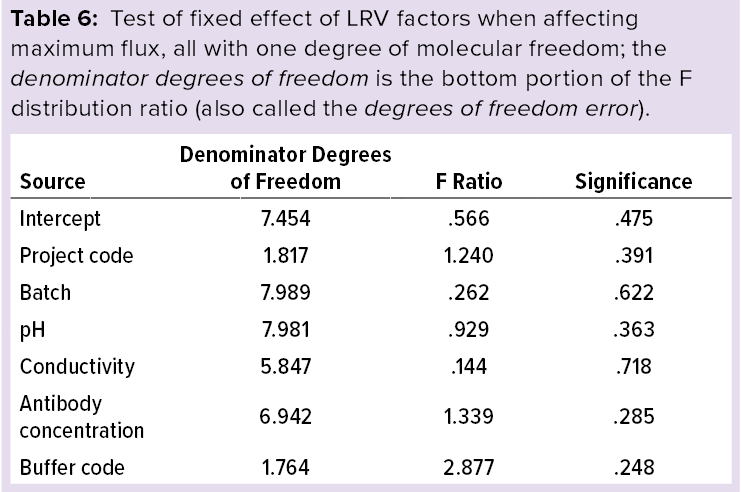
Statistical Analysis: Using IBM SPS statistics software (version 23) and a multivariate analysis model, we modeled the effects of maximum flux including pH, conductivity, and antibody concentration as covariates. Tables 5 and 6 list the statistical analysis results showing that maximum flux and LRV at the maximum flux fall within the scope of this study, and no factors significantly influence the results. Based on those results, we used the process parameters listed in the “Virus Filtration” box to evaluate the effectiveness of MAb-X nanofiltration virus removal for production of IND-stage clinical samples.
Platform-Based Virus-Control Strategy
The main purpose of preclinical research is to collect product information from production technologies, quality research, and animal studies that will pave the way for subsequent clinical trials. The preclinical stage of chemistry, manufacturing, and controls (CMC) mainly focuses on establishment of preliminary production processes and production of samples for research purposes. Under the current clinical-trial rules in China, Europe, the United States, and elsewhere, companies must determine the manufacturing process to be used in clinical production at this stage. In many cases, it is necessary to complete the preparation of clinical samples.
We adopted different viral-safety methods to address risks during clinical-lot manufacturing. First is selection and verification of production cell lines and raw materials to ensure that they do not contain infectious and pathogenic viruses; second is evaluation of the virus-removal capability of our manufacturing process; and third is determination of the appropriate process steps for testing to confirm that no infectious viruses are present in the final product solution.
Application of specific principles to remove inactivated viruses requires risk assessment of operating parameters based on scientific principles and experimental experience. Some evaluated parameters are key to virus removal, so performing them within a certain operation range can ensure the reliability of virus removal methods. Other parameters do not affect virus-removal capability. For example, temperature, pH, and time are critical to low-pH virus inactivation, but the types of inactivated protein and salt concentration are of less concern. In addition, for designing a virus-removal evaluation experiment based on understanding of the process principle, the operating parameters are set at worst-case conditions to ensure process reliability.
To evaluate the effectiveness of a virus-removal/-inactivation process, regulators usually require developers to calculate the sum of that rate for an overall manufacturing process. That should be the only way to evaluate accurately the reliability of an entire production line for future BLA submission.
Driven by regulatory guidance, our company can meet the expectations of virus clearance by applying technologies that remove or inactivate endogenous viruses and small, nonenveloped viruses to downstream processes starting from the early stages of biopharmaceutical development. In addition, integrating single-use production equipment into technology platforms provides higher manufacturing flexibility, increases the convenience and speed of operations, and eliminates the risk of residual contamination. To meet the goals of biological separation and purification, combining steps and orthogonal strategies helps us achieve the industry goals of output and quality.
Platform Substantiated
In development of the MAb-X coronavirus-neutralizing antibody, our company has adopted an established MAb production platform to produce clinical samples on a CGMP single-use production line. It has reached a good level of viral-safety control from the source and process. Based on comprehensive consideration of the development process as it relates to safety of biological products, the initial virus safety generally required for the IND stage is based on two dedicated, orthogonal steps for virus inactivation and removal.
Our present MAb manufacturing platform has undergone a wide range of tests to ensure viral safety, and no clinical reports have shown viral infection from the products under clinical study. Through analysis of our database, we have documented the reliability and robustness of this platform technology. Material and process controls (e.g., culture media, supernatant detection, and facilities) and key parameter ranges (e.g., virus-inactivation pH, time, and nanofiltration pressure and flux) are helpful and instructive in that regard.
Based on the platform experience, we deem this production process to have a reliable viral safety risk minimization capability in relation to source, process, and virus removal. Clinical-sample production conforms to the scope of the platform process, and we have concluded that virus safety is controllable for this product.
References
1 EMEA/CHMP/BWP/398498/2005. Guideline on Virus Safety Evaluation of Biotechnological Investigational Medicinal Products. European Medicines Agency: London, UK, 24 July 2008; https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-virus-safety-evaluation-biotechnological-investigational-medicinal-products_en.pdf.
2 Q5A: ICH Harmonized Tripartite Guideline on Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. US Fed. Reg. 63(185) 1998: 51074; https://database.ich.org/sites/default/files/Q5A%28R1%29%20Guideline_0.pdf.
3 Teasdale A, Elder D, Nims RW. ICH Quality Guidelines: An Implementation Guide. John Wiley & Sons: Hoboken, NJ, 2018; 312–313.
4 Ray S, Tarrach K. Virus Clearance Strategy Using a Three-Tier Orthogonal Technology Platform How to Implement a Risk-Based Approach to Eliminate Viruses Using Orthogonal Technologies. BioPharm Int. 21(9) 2008: supplement; https://www.biopharminternational.com/view/viral-clearance-strategy-using-three-tier-orthogonal-technology-platform.
5 Brorson K, et al. Bracketed Generic Inactivation of Rodent Retroviruses By Low pH Treatment for Monoclonal Antibodies and Recombinant Proteins. Biotechnol. Bioeng. 82(3) 2003: 321–329; https://doi.org/10.1002/bit.10574.
6 Mattila J, et al. Retrospective Evaluation of Low-pH Viral Inactivation and Viral Filtration Data from a Multiple Company Collaboration. PDA J. Pharmaceut. Sci. Technol. 70(3) 2016: 293–299; https://doi.org/10.5731/pdajpst.2016.006478.
7 Hamamoto Y, et al. A Novel Method for Removal of Human Immunodeficiency Virus: Filtration with Porous Polymeric Membranes. Vox Sang. 56(4) 1989: 230–236; https://doi.org/10.1111/j.1423-0410.1989.tb02034.x.
8 Parenteral Drug Association. TR41: Virus Filtration. PDA J. Pharm. Sci. Technol. 59(supplement) 2005: 8–42.
Cai Bao is vice president of biomanufacturing at Bio-Thera Solutions, Ltd., 155 Yaotianhe Street, Huangpu District, Guangzhou, Guangdong, China; 86-20-39659788; cbao@bio-thera.com; https://www.bio-thera.com. Mayank Saini is a senior production engineer, Huaqiong Wang is a MSAT process scientist, Xi Cheng is a process development engineer, and Qingfeng Dong is a statistical scientist, all at Bio-Thera Solutions, Ltd.