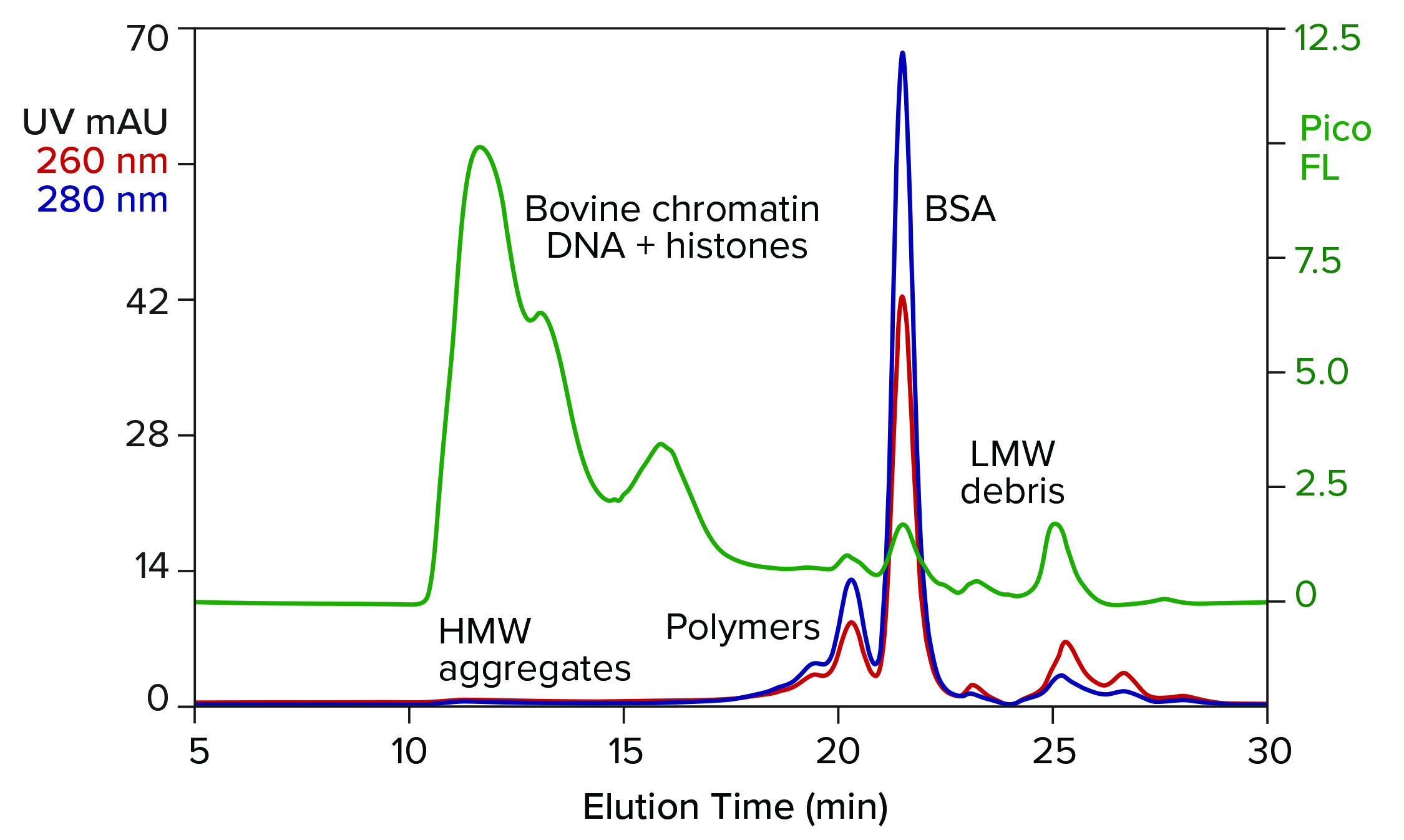
Figure 1: Analytical size-exclusion chromatography (SEC) of bovine serum albumin (BSA) marketed for processing nucleic acids; sample prestained with PicoGreen dye; TSKgel G4000SWxl, 7.8 mm × 30 cm, 50 mM MES, 150 mM NaCl, 0.05% Pluronic reagent F68, pH 6.5; flow rate: 0.5 mL/min.
A high-performing capture method is a critical bedrock asset for developing industrial purification processes. This is especially true for extended families of products that share highly similar chemical composition. Therapeutic monoclonal IgG is an example. The ability of protein A affinity chromatography to achieve 95% purity in one simple step was the runway that got recombinant immunotherapy off the ground and made it available to millions.
In fact, protein A did more. Beyond giving the industry a foundation manufacturing method, it made IgG purification accessible to immunologists without the need for advanced purification skills. It allowed them to screen efficacy, stability, pharmacokinetics, and various aspects of manufacturability for hundreds of clones, then objectively select the best qualified clinical candidates, far in advance of commercial product development.
RNA needs this kind of asset to make gene therapy and vaccines available to billions. This article shows how anion-exchange chromatography can fulfill that need.
The Reality of mRNA Purification
In vitro synthesis of RNA requires a purified DNA plasmid, purified RNA polymerase, metal-ion enzyme cofactors, and raw-material nucleotides. RNA in the completed transcription mixture is massively more abundant than the plasmid or enzymes. Purification should be simple.
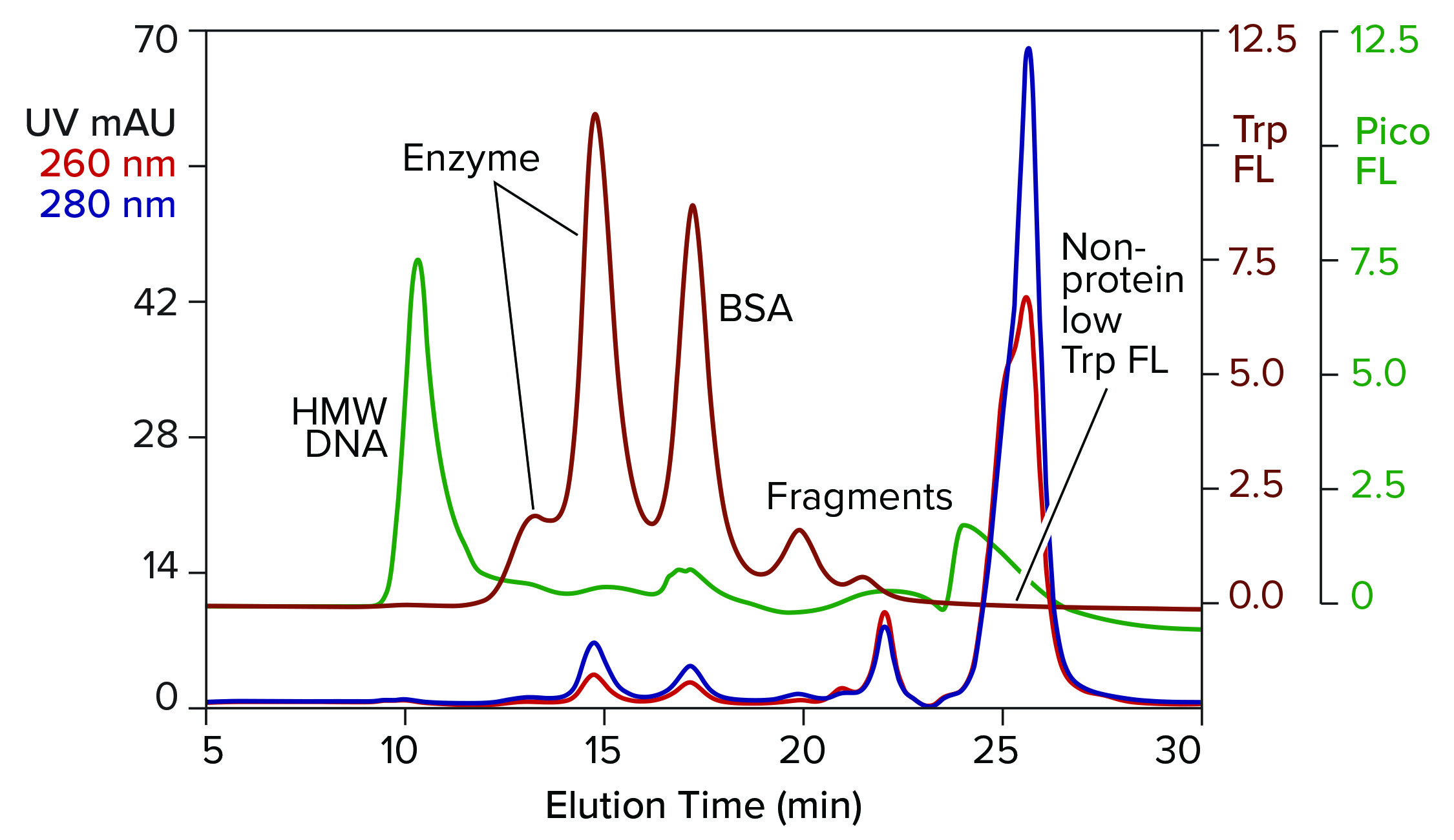
Figure 2: Analytical SEC of commercial T7 RNA polymerase prestained with PicoGreen dye; TSKgel G3000SWxl, 7.8 mm × 30 cm, 0.5 mL/min; note that the fractionation range of this column is lower than in the column of Figure 1.
But it’s not. Transcription does not always proceed to completion. In vitro transcription (IVT) mixtures commonly contain a substantial proportion of incomplete transcripts. Posttranscriptional variants include subpopulations of double-stranded RNA (dsRNA). This refers to species in which part of the sequence doubles back on itself and forms hydrogen-bonded bridges to complementary sequences on the same molecule and/or with other RNA molecules. When dsRNA is administered in vivo, recipient cells sense it as an invading virus. It must be absent from a final product to prevent triggering a toxic cytokine storm and to facilitate proper translation of the single-stranded RNA (ssRNA) (1).
Plasmids and enzymes can be sources of contaminants and adventitious agents. Plasmid preparations can be contaminated with host DNA, RNA, and proteins. Purity among commercially available polymerase enzymes is variable, and some enzymes are stabilized with bovine serum albumin (BSA). The animal origin of BSA presents a liability from the start, but it is accompanied by other contaminants that amplify the safety risk.
Figure 1 illustrates results from analytical size-exclusion chromatography (SEC) of a commercial BSA preparation marketed for enzyme stabilization in conjunction with processing nucleic acids. The assay was run on a TSKgel G4000SWxl column (Tosoh Bioscience). The intercalating fluorescent dye PicoGreen (Thermo Fisher Scientific) was added to the sample in advance to increase sensitivity of DNA detection (2). Elution was monitored with UV and fluorescence.
The most obvious feature of the chromatogram is heavy contamination by DNA. This is not pure DNA. It is the remnant of bovine chromatin: nucleosomal debris containing DNA fragments of various sizes, complexed with histones and other host proteins. Viruses tend to associate with chromatin (3–5). Endotoxins do so as well (6), and chromatin is antigenic (7, 8). The size of the DNA-protein aggregates also is a concern because high–molecular weight contributes further to their immunogenicity (9).
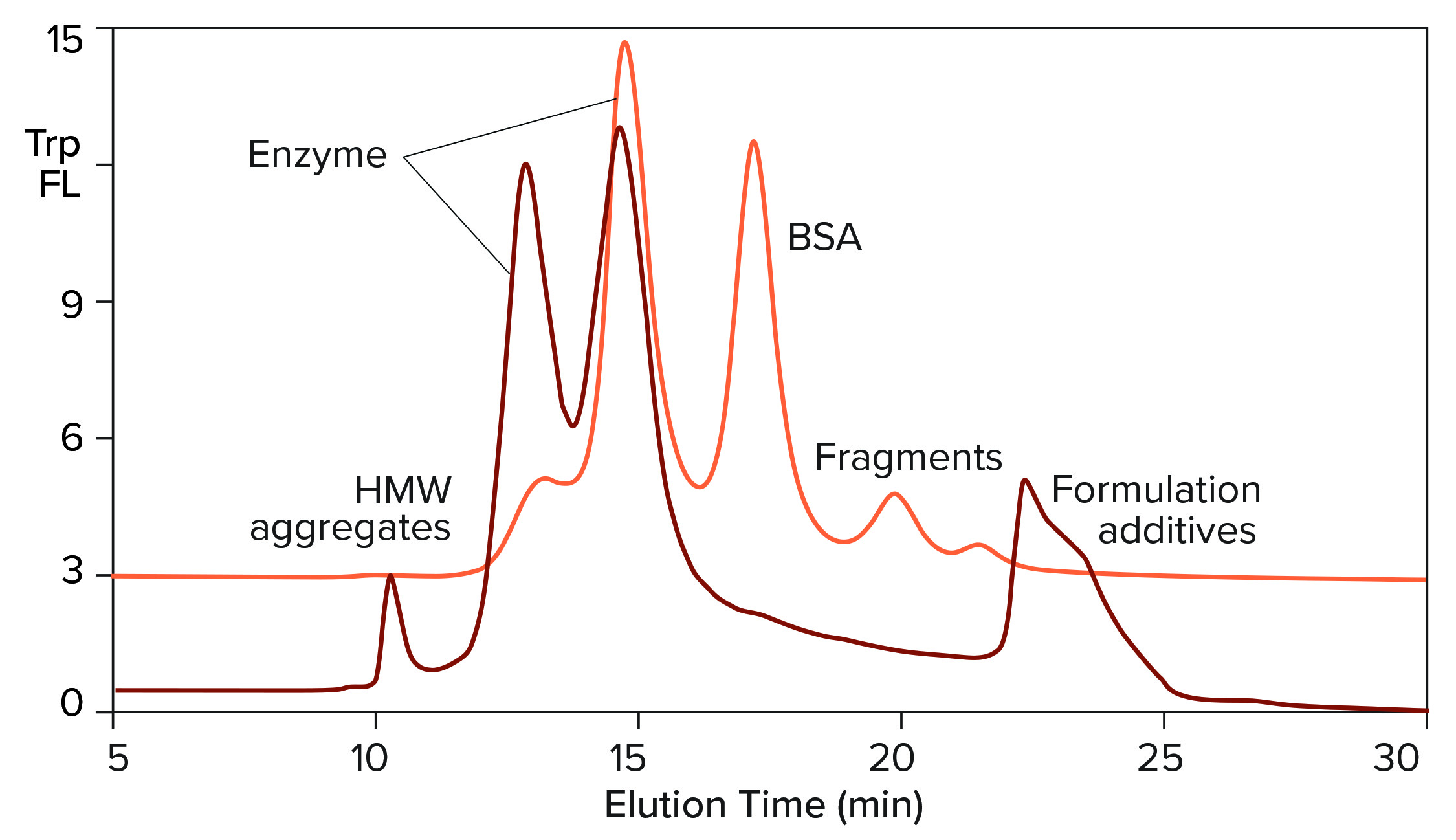
Figure 3: Overlay of SEC profiles for T7 RNA polymerase from two different suppliers; the orange profile is from Figure 2, TSKgel G3000SWxl.
The UV profile reveals high–molecular weight aggregates corresponding with the chromatin DNA. It also reveals a series of peaks on the leading side of the main BSA peak. They represent albumin homo- and hetero-polymers. A subset of albumin contains free sulfhydryl groups that enable covalent bonding to similarly endowed plasma proteins, including IgA, alpha-1 protease inhibitor, and antitrypsin-3 (10). Such protein hybrids may have a legitimate biological role in mammalian physiology, but not in IVT mixtures. Non–protein-based enzyme-stabilizing formulations are discussed elsewhere (11).
Figure 2 illustrates results from analytical SEC of a commercially available T7 RNA polymerase on a TSKgel G3000SWxl column. This single-subunit enzyme has a mass of 99 kDa. The sample was prestained with PicoGreen dye and additionally monitored for intrinsic tryptophan fluorescence. Like many enzymes, RNA polymerase is sold by the concentration of activity units. That can leave protein concentration too low for accurate monitoring by UV absorbance. Tryptophan fluorescence increases sensitivity of protein detection 15–20-fold over UV. It does not detect nucleic acids.
DNA contamination is obvious, but its source is uncertain. It could have come from the host cells used to produce the enzyme or it could have come with the BSA, or both. Aside from that, the enzyme is not homogeneous but, instead, distributed in two peaks. Heterogeneity among RNA polymerases is known (2–14). Although heterogeneity affects activity, it’s correlation with the two peaks separated by SEC is unknown.
Figure 3 overlays the tryptophan fluorescence profile of RNA polymerase from a second supplier on the profile of the first. The relative distribution of the two enzyme peaks is markedly different. Aggregate and fragment contents are higher. BSA either is absent or present in a reduced amount. This is an advantage, considering the immunogenic potential of extracellular chromatin, nonhuman proteins, and aggregates. It also leads to some practical conclusions: Enzymes in this field are not interchangeable across suppliers, and variations in enzyme heterogeneity could affect the outcome of in vitro transcription. Lot-to-lot testing is highly recommended to document quality and consistency of supply.
One more contaminant class requires consideration. The multivalent metal cations included as enzyme cofactors for transcription become contaminants in the finished IVT mixture. The phosphatidic acid residues along the ribose backbone of RNA have high affinity for such metals. RNA-metal binding can alter charge, conformation, stability, and function (Figure 4).
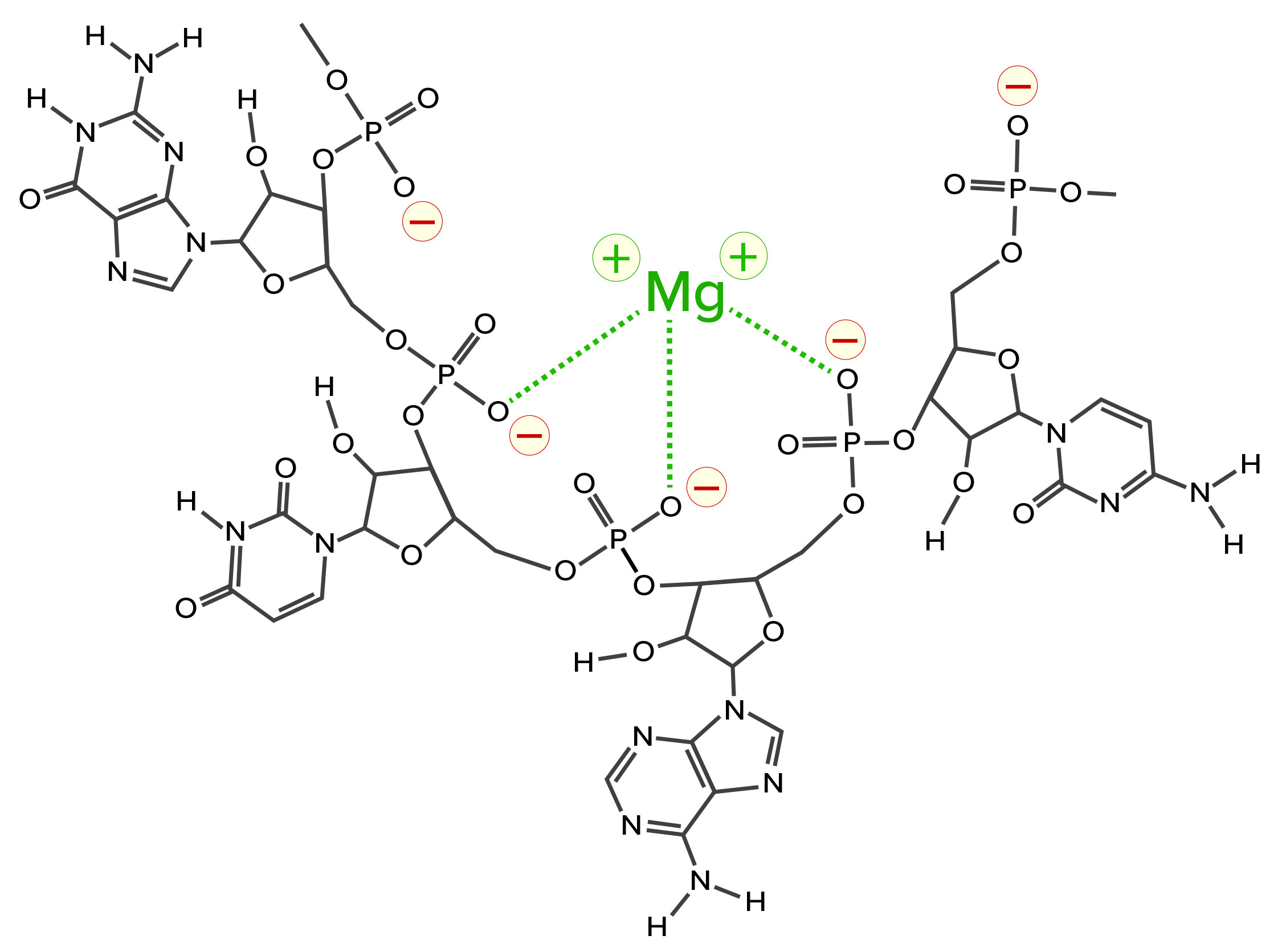
Figure 4: Coordination bonding of magnesium ions to phosphatidic acid residues on RNA; negative charges indicated in red with yellow halos and coordination bonds in green. The metal-ion positive charge can attract other RNAs and create coordination bonds with them as well.
Metal-ion coordination also has the ability to create crosslinks to other nucleic acids and proteins, forming stable intermolecular hybrids that contribute to aggregate formation. When RNA becomes the dominant species in a mixture, it outcompetes other species for the available metal ions, or it shares them in stable complexes. This is why the use of EDTA is virtually universal in chromatography buffers for purification of nucleic acids.
Figure 5 illustrates results from nanoparticle tracking analysis (NTA, using NanoSight instruments (Malvern Panalytical) of an IVT mixture containing ssRNA with a size of 1,200 bases (1.2 kb). It might be hoped that such a mixture would be dominated by a single peak. Instead, there are many peaks. Those with median size values of 37, 68, and 107 nm represent incomplete transcripts. The peak at 163 nm represents free ssRNA. The later peaks at 220 and 315 nm represent aggregates, and they contain more RNA than the apparent monomer population. To the extent that nucleic acid behavior in mammalian cell culture harvests can be trusted as a guide, these aggregates are likely to represent stable associations among RNA, DNA, proteins, and metal ions (15).
The NTA profile highlights the greatest purification challenge of all the foregoing figures. Purification is difficult enough if all the components in a sample exist as independent entities. It becomes much more difficult if contaminants are stably associated with a product. Probability of poor recovery increases in parallel. The ssRNA needs to be extracted from its associations with contaminants before it can be fractionated from them.
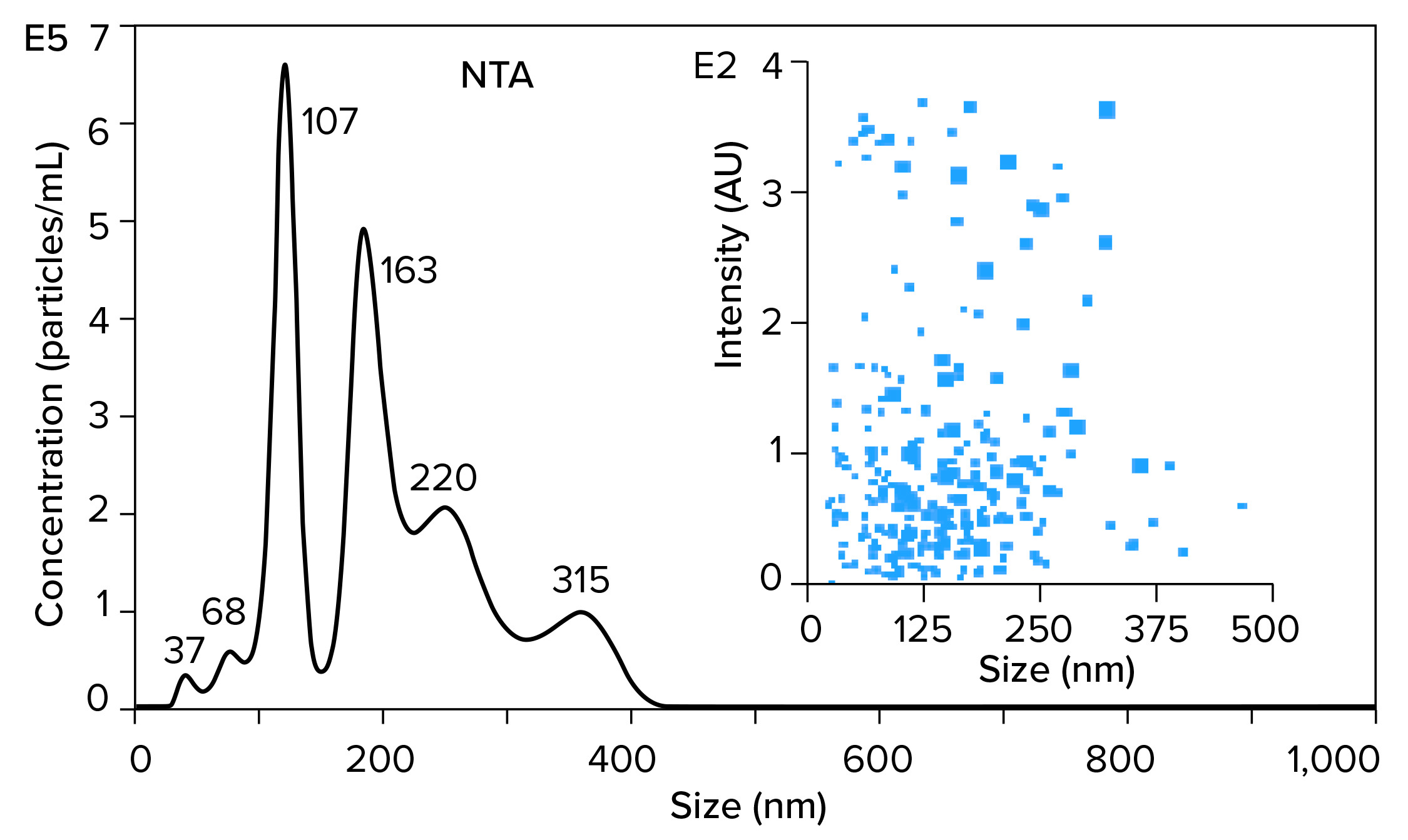
Figure 5: Nanoparticle-tracking analysis (NTA) of an in vitro transcription mixture containing 1,200 bases ssRNA
Current Tools for Industrial Purification of mRNA
An affinity method for RNA purification already exists. Hybridization-affinity chromatography with an oligo dT (poly-thymidine) ligand captures mRNA by its poly-A tail. About 250 mM sodium chloride suppresses electrostatic repulsion between the negatively charged backbone phosphatidic acid residues on the ligand and on RNA. That enables them to approach each other closely. RNA is captured by A-T base pairing. Removing the salt reestablishes their mutual charge repulsion, overwhelms A-T hydrogen bonding, and elutes the RNA (Figure 6) (11, 16, 17).
Hybridization-affinity with oligo dT can be used for capture, but it bears some limitations. It cannot discriminate ssRNA from dsRNA, and it has no ability to fractionate mRNA according to size. Intact product, incomplete transcripts, oligomers, fragments, aggregates; any species with an accessible poly-A tail elutes with all the rest. Oligo dT can be cleaned with 100 mM NaOH, but higher concentrations are not recommended. This is a concern because of the high fouling potential of IVT mixtures.
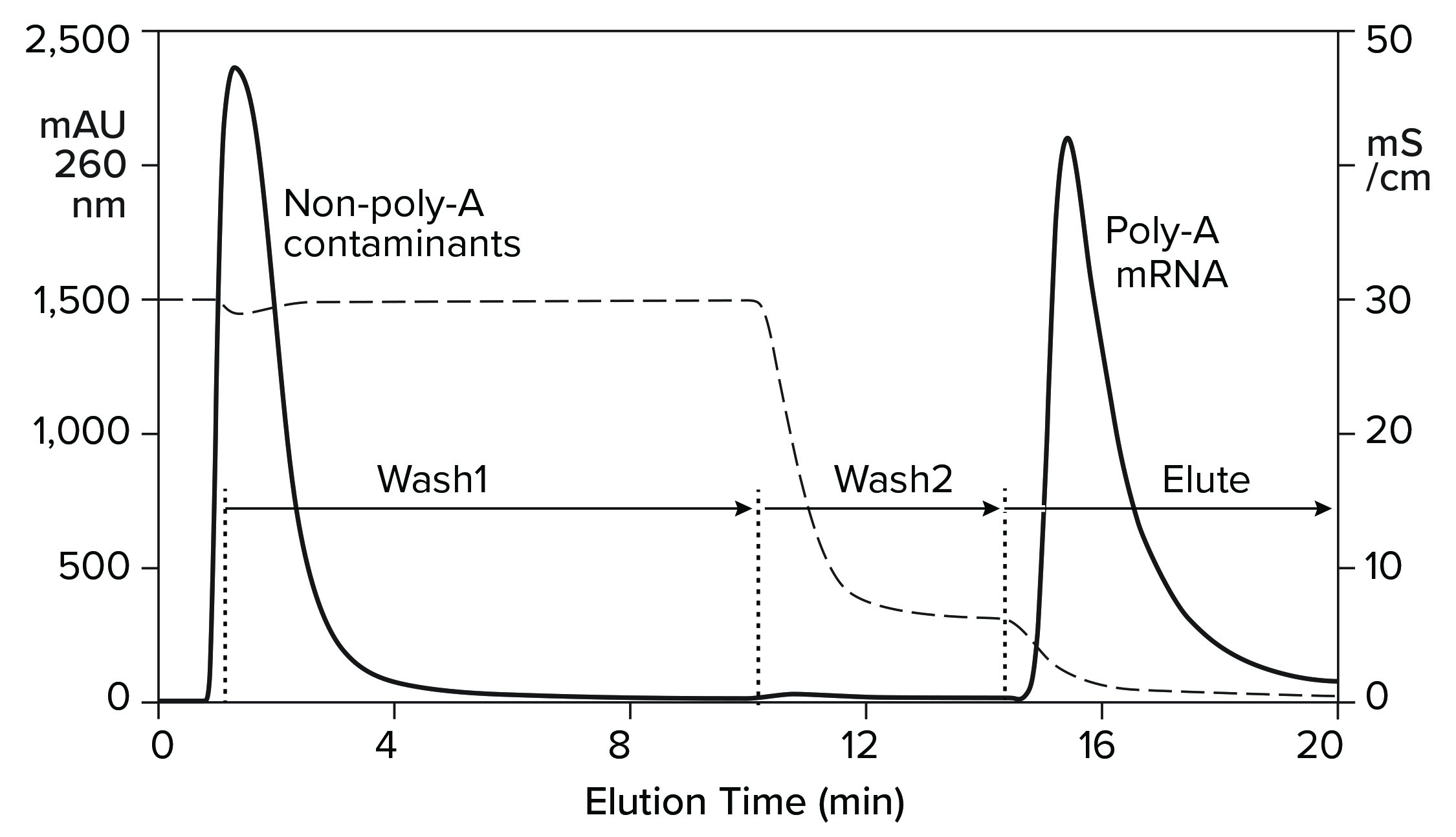
Figure 6: Hybridization-affinity chromatography of an IVT mixture on a CIMmultus Oligo dT column; equilibration/Wash1 with 50 mM sodium phosphate, 250 mM sodium chloride, 5 mM EDTA, pH 7.0; Wash2 with 50 mM sodium phosphate, 5 mM EDTA, pH 7.0;
elute = 10 mM Tris, pH 8.0.
Hydrophobic-interaction chromatography (HIC) has shown valuable utility for RNA purification. With the correct choice of binding salt, DNA and dsRNA fail to bind (Figure 7) (11). Incomplete transcripts elute before intact ssRNA, and NaOH is required to remove the majority of proteins. Reversed-phase chromatography (RPC) using styrenedivinylbenzene (SDVB) media also has proven useful to remove dsRNA and fractionate ssRNA by size (Figure 8) (18–21). HIC and RPC media are prone to fouling by crude IVT mixtures. They both tolerate extended cleaning with 1 M NaOH, so they can be restored to original condition, but fouling within a run can interfere with purification performance. This makes both of them better suited for polishing.
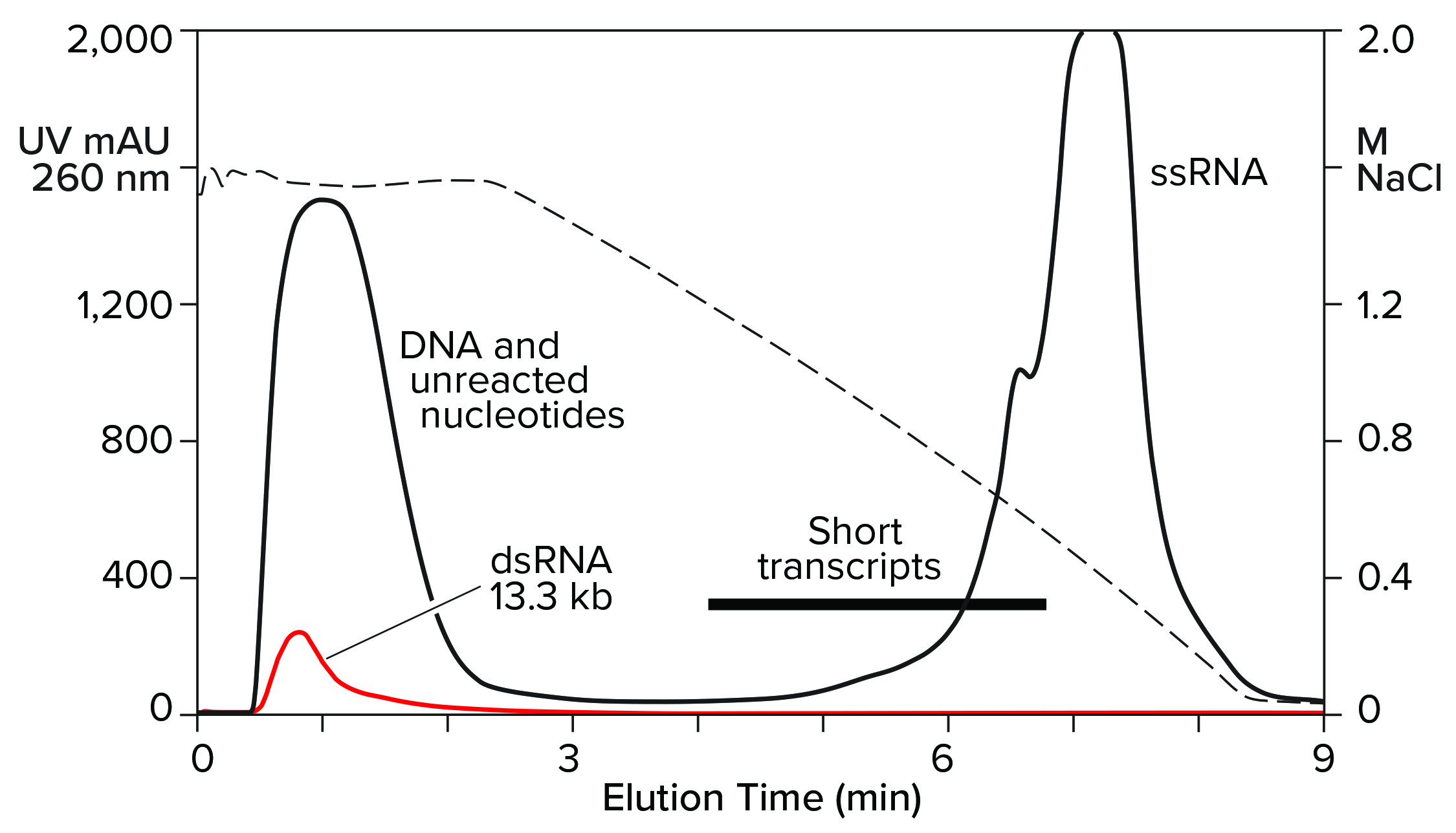
Figure 7: Hydrophobic interaction chromatography of an IVT mixture on a CIMmultus C4 HLD column; sample loaded in 1.8 M NaCl, pH 7.0, and eluted with a descending salt gradient. The red trace shows a separate dsRNA sample loaded under identical conditions.
RNA Purification by Anion-Exchange Chromatography
Ion-exchange chromatography has served protein and plasmid DNA purification for decades. Traditional exchangers such as diethylaminoethyl (DEAE) and quaternary amine (QA) work for RNA transcripts smaller than 500 bases (22, 23), but not for large transcripts. At ambient temperature, their elevated hydrogen bonding capacity prevents them from being eluted with anything less than sodium hydroxide (Figure 9). Heating the buffers and columns into the range of 50–70 °C suppresses hydrogen bonding sufficiently to enable elution with sodium chloride gradients (24).
Heating imposes a burden on process development and manufacturing, but it also provides a clue: An exchanger with less hydrogen bonding capacity should be able to elute RNA at ambient temperature. Figure 10 verifies this prediction by illustrating anion-exchange fractionation of mRNA from plasmid DNA at ambient temperature on a CIMmultus PrimaS column (BIA Separations). The sample was bound at neutral pH and eluted with an ascending pH gradient. DNA eluted before and well-separated from ssRNA. Double-stranded RNA elutes slightly after DNA but still earlier than ssRNA (11).
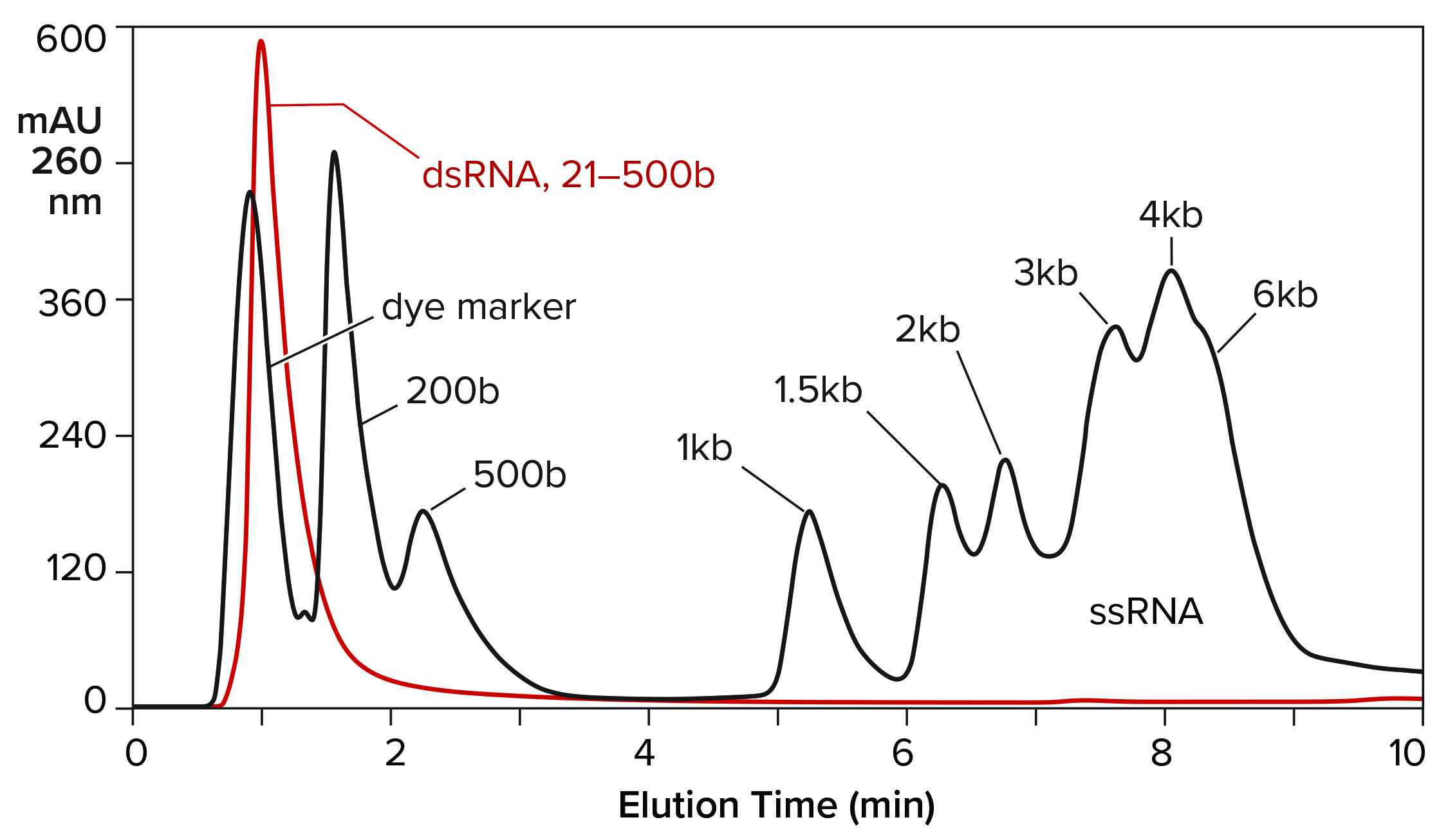
Figure 8: Reversed-phase chromatography of dsRNA and ssRNA ladders on a CIMmultus SDVB column at 65 °C; equilibration/wash buffer 0.1 M triethylaminoacetate, pH 7.0; elution buffer 0.1 M triethylaminoacetate, 25% acetonitrile, pH 7.0
Both double-stranded species can be removed instead by a neutral pH wash step with 1 M NaCl and 10 mM ethylenediaminetetraacetic acid (EDTA). Proteins are eliminated with them. Single-stranded RNA remains bound. The column is washed with another buffer to clear the excess salt, then eluted with a pH gradient. Overall purification potential of the pH gradient is increased by eliminating most of the contaminants in advance. That leaves the gradient better able to polish out their last traces. Figure 11 shows the results of this approach with the DNA plasmid and ssRNA from the previous figure. Figure 12 shows results with dsRNA and ssRNA ladders. Partial size fractionation of ssRNA is evident.
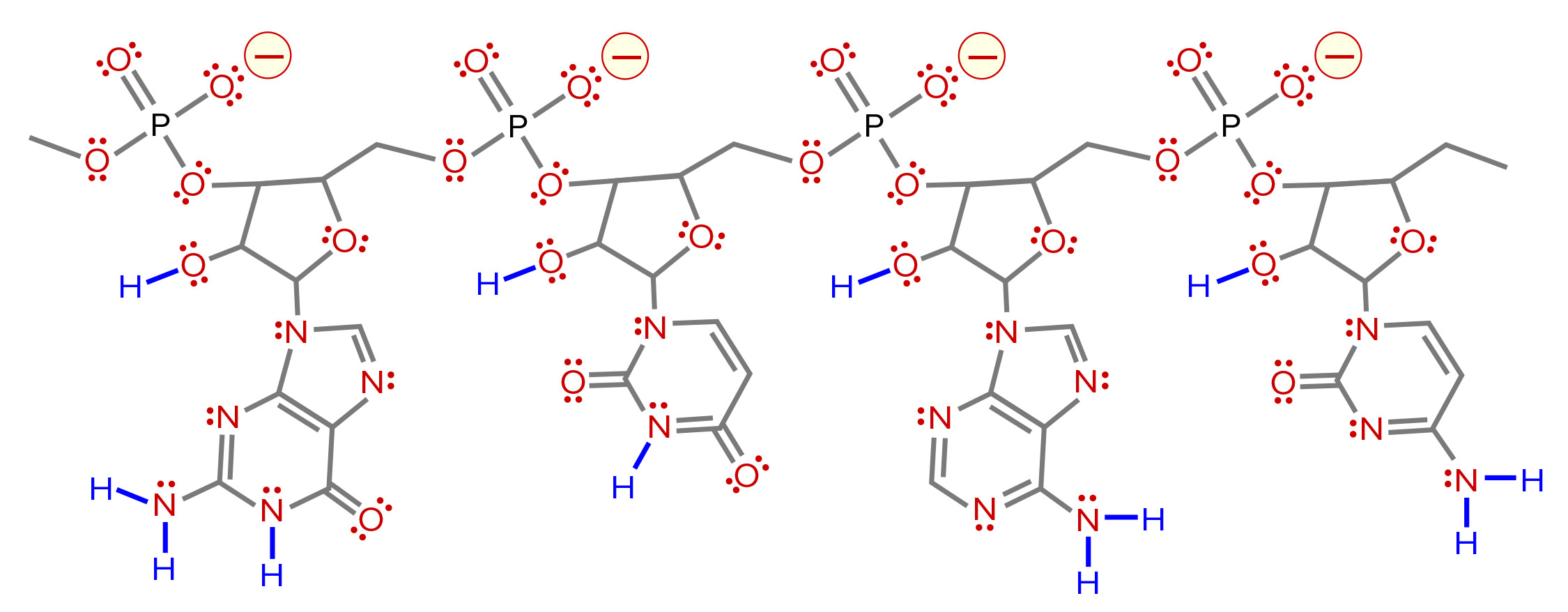
Figure 9: Distribution of charged residues, hydrogen donors, and hydrogen acceptors on RNA. Negative charges are indicated in red with yellow halos. Hydrogen acceptors are indicated in red with pairs of red dots to indicate free lone-pairs of electrons. Hydrogen donors are in blue. Compared with the four negative charges, there are 11 hydrogen donors and 75 acceptors (one acceptor for each lone pair).
The wash step can be intensified by substituting guanidine-hydrochloride for sodium chloride. Combining the chaotropic salt with EDTA simultaneously relaxes nonspecific electrostatic and hydrophobic interactions, hydrogen bonding, and metal coordination. Single-stranded RNA remains bound, which creates an opportunity to scrub and remove contaminants that may have become associated with ssRNA during transcription.
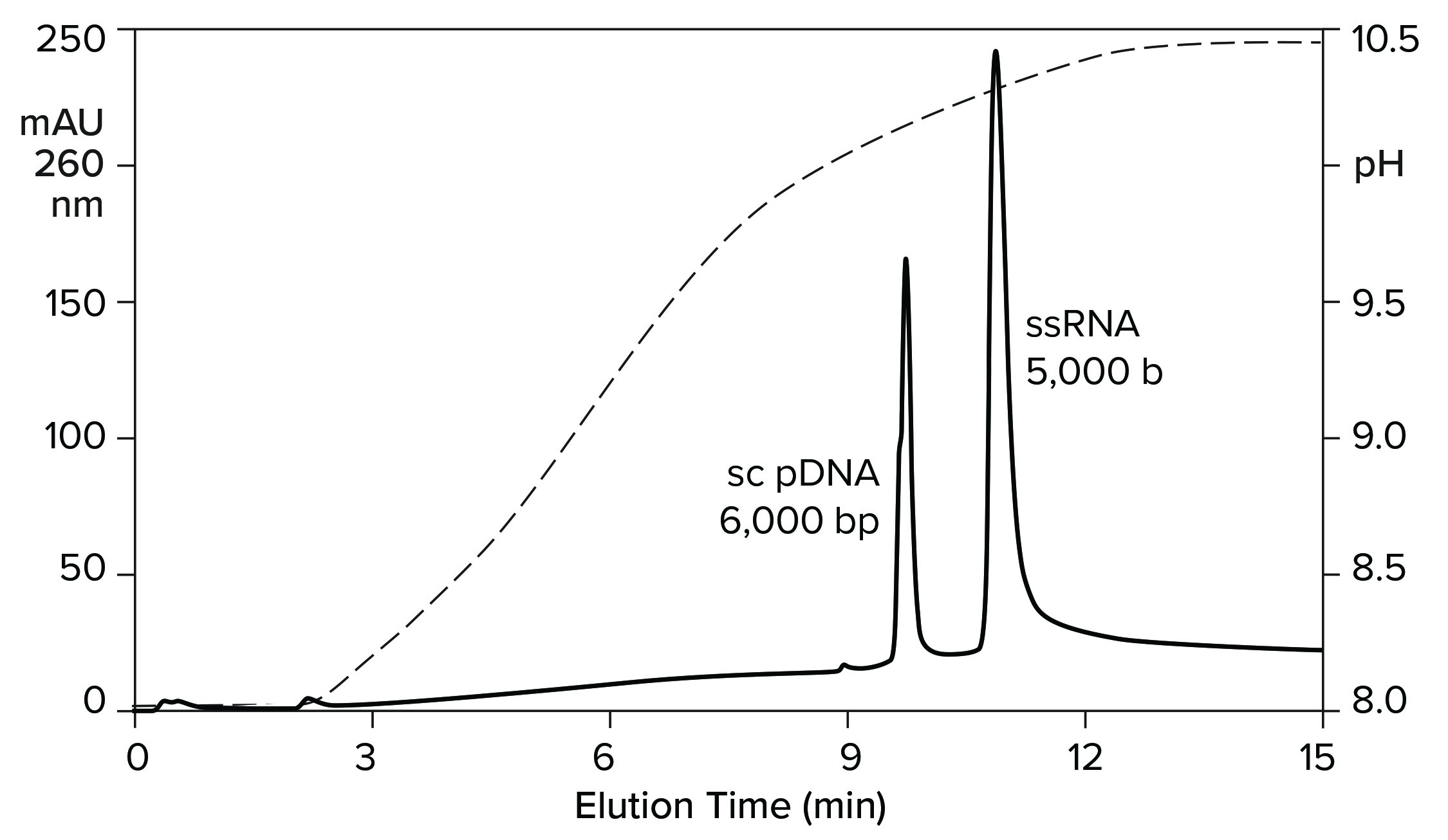
Figure 10: Anion-exchange fractionation of plasmid DNA from ssRNA by pH gradient elution on a CIMmultus PrimaS pH column; Buffer A = 20 mM Tris, 20 mM bis-tris-propane, 20 mM glycine, 50 mM NaCl, 10 mM EDTA, pH 7.0; Buffer B: 20 mM Tris, 20 mM bis-Tris-propane, 20 mM glycine, 50 mM NaCl, 10 mM EDTA, pH 11.0. Nucleic acids are from New England BioLabs.
The pH of the eluted ssRNA should be neutralized as soon as possible after elution. As in the field of protein-affinity chromatography, this is easily done by pre-aliquoting a neutralizing buffer in the fraction vessels or neutralizing the ssRNA fractions immediately after completion of the run. Brief exposure to alkaline pH produces no evidence of modification.
Like most anion exchangers, the column can be cleaned for extended periods with 1 M NaOH. Exposure to large-volume IVT mixtures generally requires treatment for at least an hour. Cleaning can be enhanced by combining 1–3 M NaCl and 10–20 mM EDTA with the NaOH. Badly fouled columns can be restored by treatment for 16–24 hours.
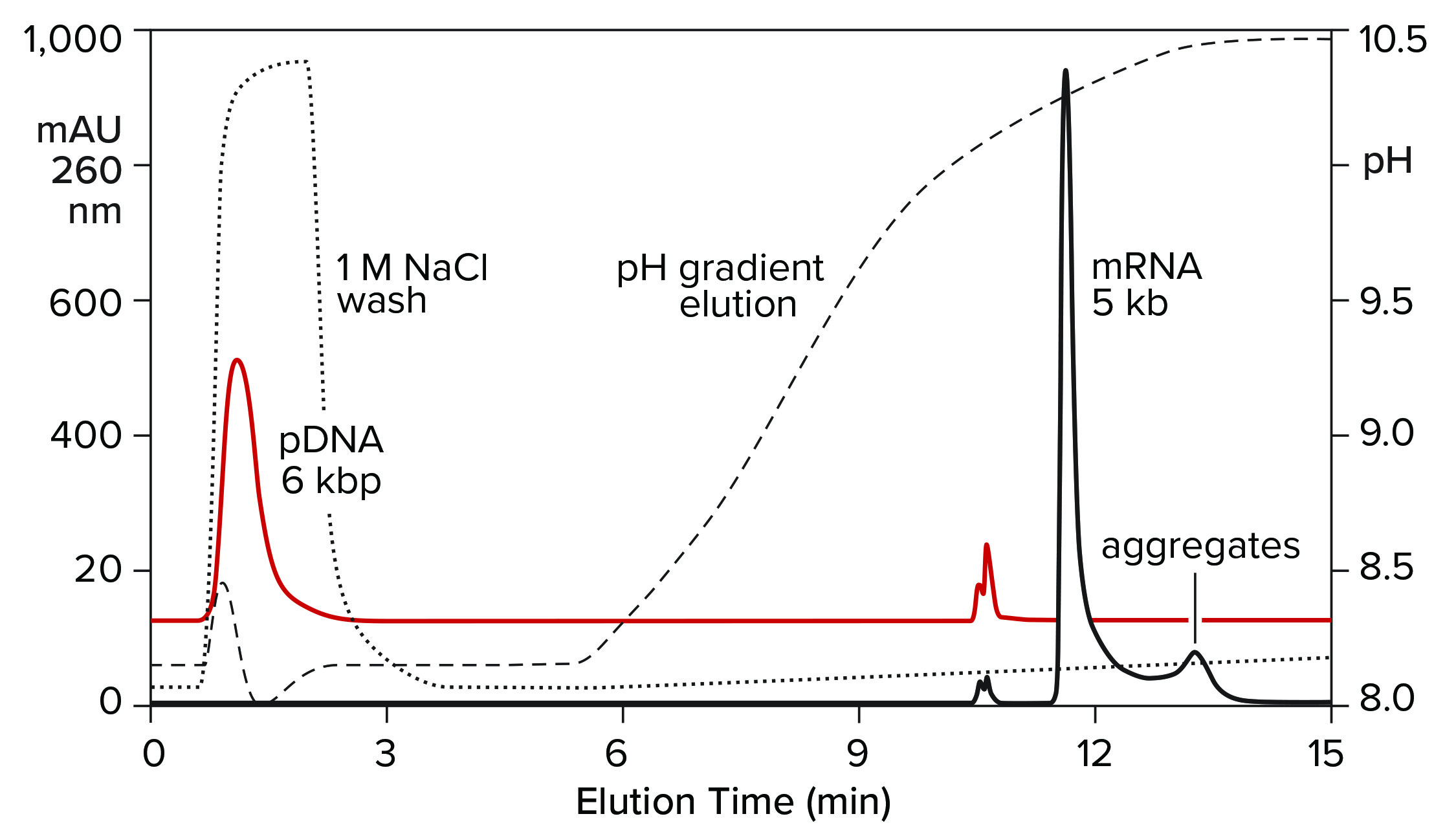
Figure 11: Elution of plasmid DNA and ssRNA from a CIMmultus PrimaS column. In one experiment, ssRNA with a size of 5,000 b was applied and eluted in a pH gradient. In the other experiment, plasmid DNA was bound then washed off the column with 1 M NaCl, 10 mM EDTA. Note its absence from the pH gradient except for a small doublet at about 11 minutes. Nucleic acids are from New England BioLabs.
Purification of Research- and Clinical-Grade ssRNA
For purification of research-quality ssRNA, a CIMmultus PrimaS column with a salt wash before pH elution provides one-step purification performance comparable to what protein A affinity has been providing to immunologists since the 1990s. This gives researchers a simple protocol to obtain small amounts of good quality ssRNA quickly and easily to advance their studies. It gives upstream process developers an easy tool to evaluate the effects of different variables in optimizing their IVT protocols.
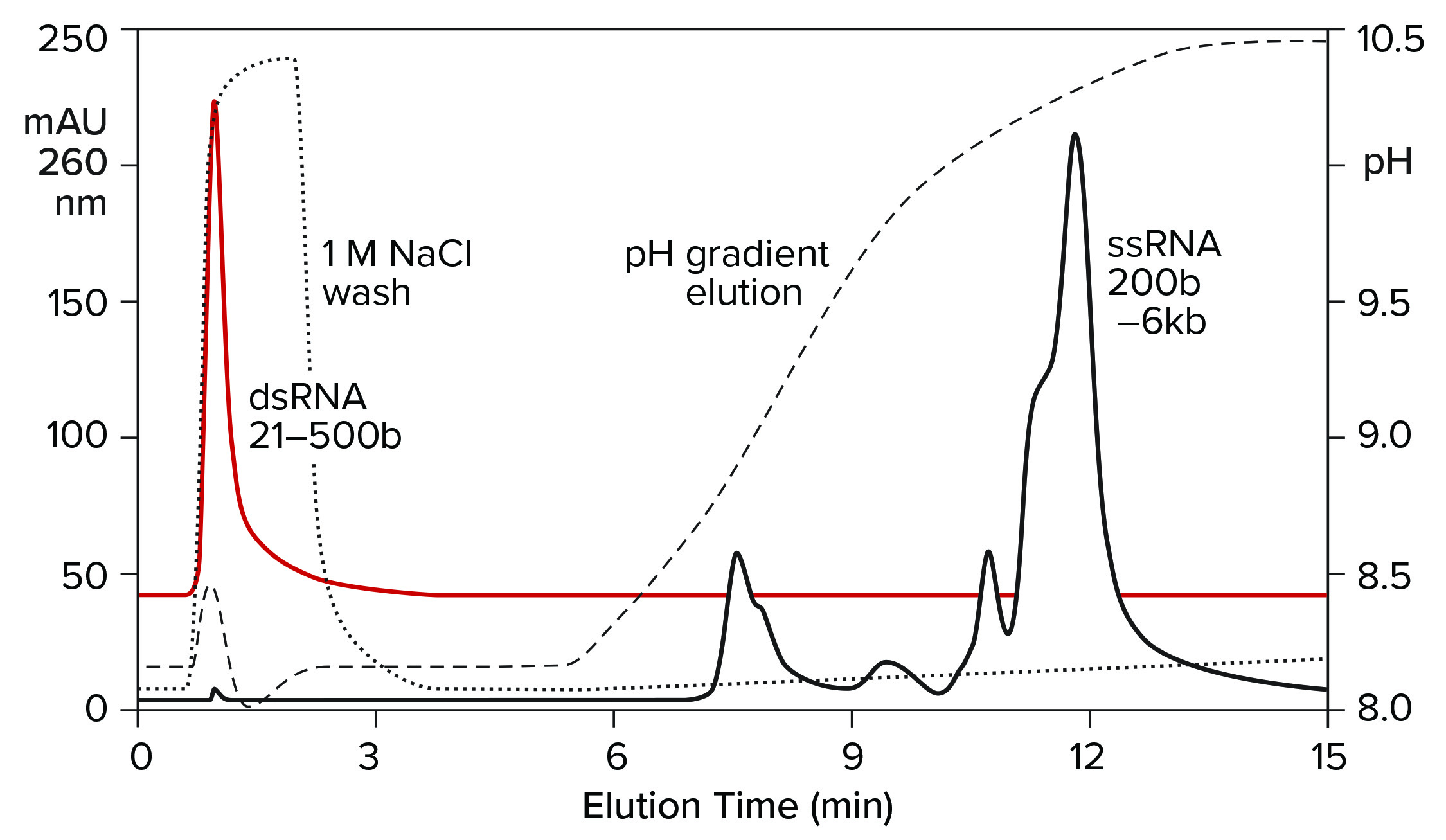
Figure 12: Elution of a dsRNA ladder and a ssRNA ladder from a CIMmultus PrimaS column. In one experiment, a ssRNA ladder containing species ranging in size from 200 to 6,000 bases was applied and eluted in a pH gradient. In the other experiment, a dsRNA ladder containing species ranging from 21 to 500 bases was bound, then washed off the column with 1 M NaCl, 10 mM EDTA. Note its absence from the pH gradient. Nucleic acids are from New England BioLabs.
It gives downstream developers the high-performing capture foundation needed for purification of clinical-quality ssRNA. It provides high initial purity and removes the foulants that could interfere with polishing methods. Polishing methods that also support removal of dsRNA, DNA, and proteins and achieve size fractionation suggest themselves as effective partners. The low salt concentration of the ssRNA coming off anion exchange enables smooth transition to RPC. It also provides a smooth workflow with HIC. Just add salt. Both platforms should consistently deliver clinical quality ssRNA.
| Vaccines and Beyond: The Future of Therapeutic RNA Beckons |
| The COVID-19 pandemic has focused the international biopharmaceutical community on the need to develop effective vaccine candidates quickly. RNA-based vaccines offer potential to accelerate development, simplify manufacturing, and lower costs compared with traditional approaches.
One of the particular benefits of RNA-based vaccines is that they represent the ultimate platform-manufacturing candidates. Chemically and structurally, they are so similar to each other that a unified development template can be applied to all of them. This lays the foundation for multivalent vaccines, for which each component can be prepared by essentially the same process. Indeed, some such products include up to six components to ensure a broad immune response to a particular pathogen. Similarity among those RNAs offers potential for manufacturing all components in a single production facility. Clinical trials already have shown efficacy against COVID-19 and other viruses including hepatitis, Zika, influenza, and rabies. More than 50 clinical trials also are proceeding using RNAs as anticancer vaccines. These properly represent immunotherapeutics rather than vaccines in the traditional sense, but they still help to illustrate RNA’s potential, and there are many therapeutic candidates Single-gene disorders in which a critical protein is not translated or is dysfunctional Clinical trials for such RNA therapies have been fast-tracked by the FDA. In the United States alone, over 30 million people suffer from rare diseases. Those tend to be under-addressed by recombinant-protein–based therapeutics because of their high development costs. The relative simplicity of developing RNA-based therapies offers hope. As in any discussion concerning the biopharmaceutical industry, antibodies eventually come into the picture, and discussion of RNA is no exception. It enables patient cells to produce their own therapeutic antibodies with absolutely authentic human glycosylation. Every therapeutic antibody presently approved for immunotherapy, every antibody currently in clinical trials, and every antibody yet to come is a potential candidate for replacement by RNA. Challenges remain, however. One challenge is delivery: getting the RNA to the site of therapy. Free RNA is broken down quickly in vivo. Incorporation into larger molecules and packaging inside liposomes or nanoparticles have shown promise. Storage is another challenge. Freezing or refrigeration currently is required. Alternatives are under development to make RNA therapies available in regions where refrigeration is unavailable. Purification represents a third challenge. Double-stranded RNA conformers and heteroaggregates of RNA with residual synthesis reagents can elicit antagonistic immune responses. Development of a purification platform that can remove those species simply, scalably, reproducibly, and with good process economy remains an important goal. In this context, COVID-19 may yet offer a positive outcome that outweighs its negative impact: By elevating the impetus to solve those remaining challenges, it can help to usher the full range of RNA-based therapies into a transformed, better prepared, and healthier new world. For Further Reading Michel T, Wendel H-P, Krajewski S. Next-Generation Therapeutics: mRNA as a Novel Therapeutic Option for Single-Gene Disorders. Modern Tools for Genetic Engineering. Kormann M, ed. IntechOpen: London, UK, 2016; https://doi.org/10.5772/62243. Pardi N, et al. mRNA Vaccines: A New Era in Vaccinology. Nat. Rev. Drug Discov. 17(4) 2018: 261–279; https://doi.org/10.1038/nrd.2017.243. RNA Vaccines: An Introduction. PHG Foundation: Cambridge, UK, 2020; https://www.phgfoundation.org/briefing/rna-vaccines. Schlake T, et al. mRNA: A Novel Avenue to Antibody Therapy? Mol. Therapy 27(4) 2019: https://doi.org/10.1016/j.ymthe.2019.03.002. Tang X, et al. Therapeutic Prospects of mRNA-Based Gene Therapy for Glioblastoma. Front. Oncol., 8 November 2019; https://doi.org/10.3389/fonc.2019.01208. Tavernier G, et al. mRNA as Gene Therapeutic: How to Control Protein Expression. J. Contr. Release 150(3) 2011: 238–247; https://doi.org/10.1016/j.jconrel.2010.10.020. The Advantages of mRNA Vaccines. Moderna Therapeutics: Cambridge, MA, 2020; https://www.modernatx.com/pipeline/therapeutic-areas/mrna-therapeutic-areas-infectious-diseases. Van Hoecke L, Roose K. How mRNA Therapeutics Are Entering the Monoclonal Antibody Field. J. Transl. Med. 17, 2019: 54; https://doi.org/10.1186/s12967-019-1804-8. |
Acknowledgments
Some of the figures in this article were adapted from 11, with permission from the publisher.
References
1 Gantier M, Williams B. The Response of Mammalian Cells to Double-Stranded RNA. Cytokine Growth Factor Rev. 18(5–6) 2007: 363–371; https://doi.org/10.1016/j.cytogfr.2007.06.016.
2 Tan L, et al. Characterization of DNA in Cell Culture Supernatant By Fluorescence-Detection Size-Exclusion Chromatography. Anal. Bioanal. Chem. 407(14) 2015: 4173–4181; https://doi.org/10.1007/s00216-015-8639-9.
3 McPhillips M, Ozato K, McBride A. Interaction of Bovine Papillomavirus E2 Protein with Brd4 Stabilizes Its Association with Chromatin. J. Virol. 79(14) 2005: 8920–8932; https://dx.doi.org/10.1128%2FJVI.79.14.8920-8932.2005.
4 Knipe D, et al. Snapshots: Chromatin Control of Viral Infection. Virol. 435(1) 2013: 141–156; https://dx.doi.org/10.1016%2Fj.virol.2012.09.023.
5 Lieberman P. Chromatin Regulation of Virus Infection. Trends Microbiol. 14(3) 2006: 132–140; https://doi.org/10.1016/j.tim.2006.01.001.
6 Augusto L, et al. Histones: A Novel Class of Liposaccharide-Binding Molecules. Biochem. 42(13) 2003: 3929–3938; https://doi.org/10.1021/bi0268394.
7 Goldblatt D, Bustin M. Antigenicity of Histones in Various Chromatins. Biochim. Biophys. Acta 606(2) 1980: 304–315; https://doi.org/10.1016/0005-2787(80)90040-4.
8 Goldblatt D, Bustin M. Exposure of Histone Antigenic Determinants in Chromatin. Biochem. 14(8) 1975: 1689–1695; https://doi.org/10.1021/bi00679a022.
9 Rosenberg A. Effects of Protein Aggregates: An Immunologic Perspective. AAPS J. 8(3) 2006: E501–507; https://doi.org/10.1208/aapsj080359.
10 Schultze HE, Heremans JF. Molecular Biology of Human Proteins with Special Reference to Plasma Proteins, Vol. 1: Nature and Metabolism of Extracellular Proteins. Elsevier: Amsterdam, The Netherlands, 1966.
11 Gagnon P. Purification of Nucleic Acids: A Handbook for Purification of Plasmid DNA and mRNA for Gene Therapy and Vaccines. BIA Separations: Ajdovščina, Slovenia, 2020: https://www.biaseparations.com/en/products/monolithic-columns/books.
12 Coban O, et al. Conformational Heterogeneity in RNA Polymerase Observed By Single-Pair FRET Microscopy. Biophys. J. 90(12) 2006: 4605–4617; https://dx.doi.org/10.1529%2Fbiophysj.105.078840.
13 Fukuda R, Iwakura Y, Ishihama A. Heterogeneity of RNA Polymerase in Escherichia coli: A New Holoenzyme Containing a New Sigma Factor. J Mol Biol. 83(3) 1974: 361–367; https://doi.org/10.1016/0022-2836(74)90284-8.
14 Westpheling J, Ranes M, Losick R. RNA Polymerase Heterogeneity in Streptomyces coelicolor. Nature 313, 3 January 1985: 22–27; https://doi.org/10.1038/313022a0.
15 Gagnon P, et al. Nonspecific Interactions of Chromatin with Immunoglobulin G and Protein A, and Their Impact on Purification. J. Chromat. A. 1340, 2014: 68–78; https://doi.org/10.1016/j.chroma.2014.03.010.
16 Satterfield B, et al. Microfluidic Purification and Preconcentration of mRNA By Flow-Through Polymeric Monolith. Anal. Chem. 79(16) 2007: 6230–6235; https://doi.org/10.1021/ac0709201.
17 Slater R. The Purification of Poly(A)-Containing RNA By Affinity Chromatography. Meth. Molec. Biol. 2, 1984: 117–120; https://doi.org/10.1385/0-89603-064-4:117.
18 Karikó K, et al. Generating the Optimal mRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding mRNA. Nucleic Acid Res. 39(21) 2011: e142; https://doi.org/10.1093/nar/gkr695.
19 Weismann D, et al. HPLC Purification of In Vitro Transcribed Long RNA. Meth. Molec. Biol. 969, 2013: 43–54; https://doi.org/10.1007/978-1-62703-260-5_3.
20 Azarani A, Hecker KH. RNA Analysis By Ion-Pair Reversed-Phase High Performance Liquid Chromatography. Nucleic Acid Res. 29(2) 2001: e7; https://dx.doi.org/10.1093/nar/29.2.e7.
21 Majors RE. The Cleaning and Regeneration of Reversed-Phase HPLC Columns. LC-GC Europe 21(1) July 2003: 19–26; https://cdn.sanity.io/files/0vv8moc6/chroma/1ab2a45ce4f7c16aa08f343c168ed8a12a4aa.pdf.
22 Prazeres D, Schluep T, Coony C. Preparative Purification of Supercoiled Plasmid DNA Using Anion Exchange Chromatography. J. Chromatog. A 806(1) 1998: 31–45; https://doi.org/10.1016/s0021-9673(97)01254-5.
23 Koubek J, et al. Strong Anion-Exchange Fast Performance Liquid Chromatography as a Versatile Tool for Preparation and Purification of RNA Produced By In Vitro Transcription. RNA 19(10) 2013: 1449–1459; https://doi.org/10.1261/rna.038117.113.
24 World Patent Application WO2014144767. Ion Exchange Purification of mRNA. WIPO: Geneva, Switzerland, 15 March 2013; https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014144767.
Corresponding author Pete Gagnon is chief scientific officer at BIA Separations, Ajdovščina, Slovenia, and a member of BPI’s Editorial Advisory Board; pete.gagnon@biaseparations.com. Špela Peršič and Blaž Goričar are members of the DNA plasmid and mRNA purification process development team, and Urh Černigoj is the head of new product development. Aleš Štrancar is chief executive officer, BIA Separations.