
STORYBLOCKS (WWW.STORYBLOCKS.COM)
Even with increased understanding of host cell proteins (HCPs) and their potential risks, no practical approach has been made available for HCP risk management during bioprocess development. A BioPhorum Development Group (BPDG) team has identified common HCP-related risk factors and built a template for semiquantitative risk assessment during process development based on publicly available information. To this end, the BPDG HCP working team’s assay and knowledge-sharing experts have established a common HCP risk assessment tool and mitigation strategy to guide bioprocess developers. In part 1 last month, we introduced this tool and described how to apply it when high levels of residual HCPs are found in a drug substance (DS). This month, we examine several other applications as well as address the limitations of our risk-assessment tool.
More Potential Negative Events
Dilutional Nonlinearity Issues with Sample Testing: In measurements of residual HCP in drug substance or in-process samples, dilutional linearity assessment is essential to ensure accurate determination of trace HCPs in a complex sample matrix dominated by the presence of drug product (5, 27).
Enzyme-linked immunosorbent assays (ELISAs) are multianalyte assays involving many antibody–antigen immune reactions. The signal generated from the sum of all antibody–antigen reactions in a sample is calibrated against a curve plotted with signals generated from a standard series against a known total protein concentration. Because of HCP composition differences between samples and standards, dilutional linearity often is limited to a narrow range in which the signal response proportionally correlates with the residual HCP amount. Dilutional nonlinearity often is observed when an insufficient amount of anti-HCP antibody is available to capture all highly abundant HCPs that copurify with DS.
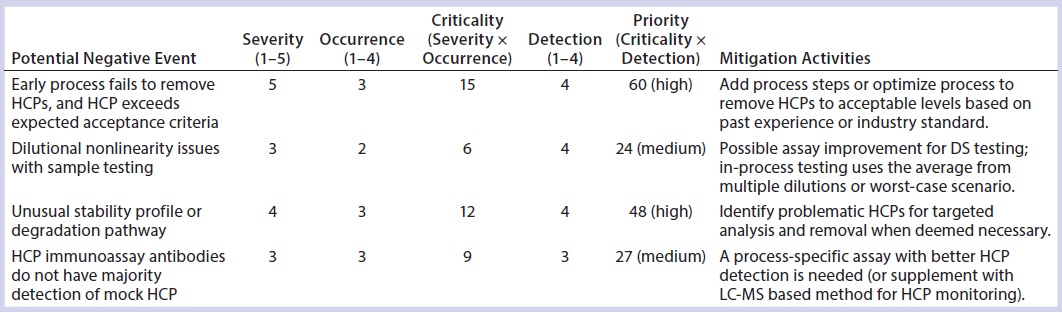
Table 8: Host-cell protein (HCP) risk scoring and control for the most common negative events that occur during bioprocess development
Troubleshooting the potential cause for dilutional nonlinearity issues often requires the use of orthogonal methods to identify and characterize HCPs. One example of such a nonlinearity issue led to the identification of PLBL2 in CHO-produced monoclonal antibody products (27). Highlighting a common observation seen during HCP assay qualification, Table 8 lists the potential risks from dilutional nonlinearity with a severity score of 3, occurrence score of 2, and the risk criticality of 6 (3 × 2). The detectability of its impact is considered to be very difficult, with a detection score of 4. Therefore, the risk priority number (RPN) is evaluated to be 24 (3 × 2 × 4), which falls into the medium-risk range (21–44). No process change may be necessary in such a case if the HCP amount measured also falls within the low-risk range, but assay changes or orthogonal methods might be needed to control the risks (Table 8).
A more accurate assessment can be made in some cases depending on the available analytical data. As recommended by USP monograph chapter 1132, when dilution linearity cannot be achieved even after assay improvement and analytical investigation, DS testing results can be reported using the worst-case scenario. In such cases, an analyst reports the HCP amount determined from a DS dilution that gives the highest level of parts-per-million.
Products with Unusual Stability Profiles or Degradation Pathways: The effect of residual HCP on product stability often is unknown until DS stability data become available. Residual HCPs may have protease activity that can degrade a therapeutic protein, leading to unusual stability profiles with unexpectedly high levels of fragmentation, for example (22, 28). Some HCPs may have lipase activity that can degrade polysorbates used in formulations, and that can lead to formation of visible or subvisible particles in drug products (8, 20, 24, 29). Degradation of polysorbate also can decrease product quality through promotion of methionine oxidation and aggregation. That in turn can create a potential immunogenicity issue.
Polysorbate often is added in excess to compensate for its degradation over time. However, subvisible or visible particle formation resulting from that degradation can cause significant product quality concerns. In such cases, the severity is considered to be significant, with a score of 4, and the product quality failure is probable, with an occurrence score of 3, leading to a risk criticality score of 12 (4 × 3). Detectability is difficult, with a score of 3, given the time required to acquire meaningful product stability data and the need to identify the cause of unusual stability profiles. Altogether, the RPN is 36 (4 × 3 × 3), which falls within the medium risk range, for which further analytical characterization to identify problematic HCPs would be recommended (Table 8). The risk of such a negative event can be reassessed upon identification of problematic HCPs and targeted analysis of their levels and enzymatic activities. When reassessment results in a higher RPN number (≥45), process improvements might be needed to further remove such HCPs to acceptable levels.
Other Factors to Consider When Assessing HCP-Related Risk: Biologics development is a continuous process through which product and process understanding builds up with extensive analytical characterization. That in turn helps guide process improvements to deliver a final product with high quality in terms of purity, safety, and efficacy. During this dynamic process, risk priorities change frequently along with risk severity, occurrence, and detection. Often they need to be reevaluated to identify proper mitigation activities. Companies can apply our risk-assessment methodology to their own unique sets of circumstances to manage and minimize potential risks posed by HCPs.
In addition to improvements in downstream processing, it is also important to adopt relevant and robust analytical assays for HCP measurement and characterization. Therefore, these same principles also can apply in evaluating the risk priority for new assay development or orthogonal approaches for HCP characterization. Table 8 also lists some of the most common negative events observed with HCP assays, with risk assessment and mitigation activities.
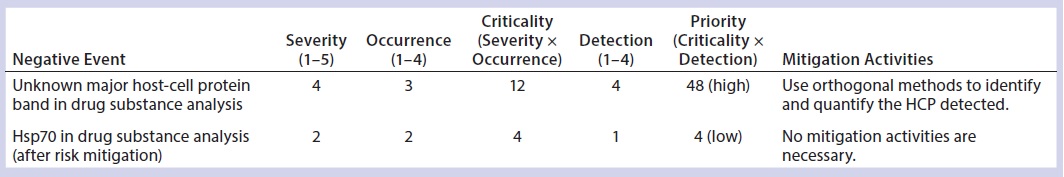
Table 9: Heat-shock protein 70 (Hsp70) case study (30)
Real-Life Industry Case Study: Ideally, risk-assessment tools should be living documents that are continually reevaluated throughout product and process development. An example of such a reevaluation process can be found in a recent HCP investigation by Lu, et. al (30). In their study of a recombinant fusion protein expressed in human embryonic kidney (HEK) cells and purified with a three-column process, one major HCP band was observed consistently in DS samples after that three-step purification. The team’s initial assessment of risk using the BPDG risk-assessment tool yielded a high risk priority with a cumulative score of 48 (4 × 3 × 4). Before the HCP was identified, it was given a severity of 4 because of both high uncertainty and its unknown status.
One-dimensional (1D) gel electrophoresis using anti-HEK HCP antibodies confirmed the presence of one major HCP. That one major HCP would severely compromise safety is probable, with an occurrence score of 3. Detectability of this particular HCP’s impact was considered to be very difficult without identification information, giving it a detection score of 4. With such a high priority score, orthogonal tools would be needed to identify and quantify the HCP to decrease its risk profile.
Liquid chromatography with mass spectrometry (LC–MS) identified the HCP as heat-shock protein 70 (Hsp70), which was confirmed using other analytical methods (31). Hsp70 is a chaperone protein found in mammalian cells and released on cell breakage. It is a common HCP identified in bioprocesses (31), ubiquitously expressed in human tissues and thus present in human plasma. The amount of human Hsp70 introduced through administration of a recombinant fusion protein would be negligible and far below the levels of any stress-related Hsp70 release, so it should pose minimal risk to patients.
After the HCP was identified, quantified, and deemed to be safe for the administered dosing, its risk priority was reevaluated. With a better-defined clinical risk, the severity score was decreased to 2; the occurrence score also decreased to 2. Finally, because the HCP had been identified and specific assays developed to measure its presence and quantity, the detectability score was lowered to 1, leading to a final RPN of 4 (2 × 2 × 1). With such a low priority score, no further process or assay changes were required.
This case study illustrates both how the risk-assessment tool can be used to identify the need to tackle experimentally unknown HCPs in DS samples and the benefits of using it as a living document. This example also highlights the necessity of using both our tool and a clinical safety risk assessment tool as described by de Zafra, et al. (7) to decrease the risk profile of a contaminating HCP.
Further Discussion
Measurement, control, and potential risks of residual HCPs in biologics are well discussed in literature and regulatory guidelines (1, 5, 10, 11). The risks associated with such impurities often are assessed with a combination of downstream process capabilities, residual HCP levels, the (maximum) dose used, route of administration, dosing frequency, toxicological data, clinical indication, and clinical data (1, 7). Although it is a common understanding that HCPs pose some clinical safety risk through their potential to elicit an immune response, only a few clinical experiences indicate a clear correlation of residual HCP existence and antidrug antibody or anti-HCP antibody response in patients. It is difficult to demonstrate which HCP at what concentration can cause immunogenicity problems in humans under different disease conditions (1).
Theoretically, preclinical pharmacological and toxicological evaluations can be performed with different amounts of HCP impurities present. However, most such evaluation results are irrelevant because of the differences in immunogenicity responses to HCP impurities between animal models and human patients. The magnitude and nature of an immune response depends on homology of an HCP’s amino acid sequence, the residual HCP amount, a product’s dosing regimen, a patient’s health condition, and so on. For that reason, a risk-control strategy must be applied through development of robust downstream processing steps to remove HCPs (1, 5, 7).
In addition to eliminating HCPs, downstream processing also needs to remove other process-related impurities such as host-cell DNA, residual chromatography ligands, and adventitious viruses. Some platform purification processes designed for a class of molecules can be incapable of addressing certain difficult-to-remove HCPs that copurify with a DS because of their high abundance in cell culture supernatant, similar biochemical properties with the DS (product nonspecific), or hitchhiking effects (product specific) (15, 32–35). Although increasing the number of column washes and adding additional purification steps have been shown to remove HCPs that nonspecifically copurify with DS to lower levels (37), those additional steps will increase costs in biologics production. Thus, it is expected that every biological drug product will have trace amounts of HCPs residing with the drug substance.
Setting tolerance levels of residual HCPs in biologics is a balance of process capability and manufacturing cost on one side and potential safety risk on the other. So HCP risk assessment is an inherent part of bioprocess development, from preclinical studies all the way through commercialization. The goal is for a purification process to minimize HCPs residing with a DS and for process characterization to elucidate and monitor their clearance during downstream processing. With a robust downstream process and sensitive analytical methods to characterize HCPs during process development, the risks associated with HCPs should be minimized to low levels by late-phase development.
When a manufacturing process is fixed for commercial production, the risk of residual HCPs in drug product to patient safety can be assessed following the framework published by de Zafra et al. (7). However, practical risk assessment guidance has been lacking for bioprocess developers that haven’t yet identified residual HCPs in their drug substances. The riskassessment tool developed through the BPDG collaboration adopts a quality risk-management concept as described in the ICH Q9 guideline (23). Our aim has been to develop a tool that could be used semiquantitatively to calculate risks associated with residual HCPs based on common HCP-related negative events encountered during biologics development.
This tool, however, comes with a few limitations. Severity, occurrence, and detection scores may be considered subjective. Negative events defined herein don’t reflect all information available at the time of risk assessment, so the risk scores presented in our examples or templates should be reevaluated when new information is uncovered. The risk severity, occurrence, and detection as defined in Table 1 (see Part 1 last month) is to evaluate the risk on the impact of a negative event when it occurs. Those definitions are different from the probability of occurrence of a given negative event and the detectability of negative event, which proactively evaluates the risk of a negative event.
Aside from those limitations, our risk-assessment tool provides a template for semiquantitative risk assessment in which a negative event can be defined further based on a given company’s unique circumstances. By applying this tool, risk priority can be identified so mitigation actions can be taken to minimize potential risks caused by HCPs in subsequent process development. Such risk-evaluation and mitigation activities should help companies communicate with regulatory agencies regarding steps they have taken to minimize process risk and defend their decisions not to add additional process steps or control measures when the assessment implies that such measures are unnecessary.
A Strategic Approach
With guidance absent on HCP-related risk management during product development, we determined that a risk-assessment tool to guide product risk control would be of particular interest to the biopharmaceutical industry. By developing the tool described herein, we hope to establish an industry-wide best practice through the BioPhorum Development Group’s HCP collaboration discussions. This risk-management approach could be used to mitigate risks of introducing HCPs that might adversely affect clinical product safety or efficacy or product stability in early and late-stage process development. It also can be used to identify the risks of HCPs already present in a DS and develop risk mitigation strategies if needed.
The purpose of this tool is to provide a strategy that can be applied consistently to products in a company’s development pipeline during process and assay development and process/assay changes. This tool combines science and risk-based approaches to mitigate HCP risk in products. It can be refined to better reflect the multiple negative events that affect product purity and quality and patient safety. Companies adopting this tool can apply their own circumstances, products, and experiences to a well-defined risk evaluation the product-development life cycle and use the tool to prioritize their risk mitigation activities as needed during process or assay development. In addition, the same methodology can be applied to assess safety risks from individual HCPs identified by LC–MS characterization of residual HCPs in drug products, taking multiple clinical risk factors into consideration. The main advantage of this tool is to provide semiquantitative assessment of risk and allow input from different subject-matter experts to help manage risks and facilitate a decision-making process.
You can download a more detailed risk-assessment tool developed by the BioPhorum Development group here: www.biophorum.com/host-cell-protein-risk-assessment-tool. It includes an example template that users can download apply it to their own companies’ internal processes/procedures and product specifics.
Acknowledgments
Here we describe a consensus view from the BioPhorum Development Group’s (BPDG’s) Host-Cell Protein (HCP) working team on a risk-assessment methodology used for HCP risk management and control during bioprocess development. We sincerely thank the following members of the working team from for their contribution at monthly BPDG discussions and during preparation of this manuscript: Taro Fujimori and Karen Hurkmans (AbbVie Bioresearch Center in Worcester, MA); Nesredin Mussa (Bristol-Myers Squibb in Devens, MA); Marisa Jones (GSK in King of Prussia, PA); Jason Crosby (ImmunoGen, Inc. in Waltham, MA); Yuichi Minato and Yasuyoshi Chihara (Kyowa Hakko Kirin Co., Ltd. in Gunma, Japan); Edward Rocnik (Takeda Pharmaceuticals in Cambridge, MA); David Streelman and Aniello Dan Pennetti (Eisai Morphotek, Inc. in Exton, PA); Jeffrey M. Schneiderheinze, Jamie C. Rusconi, and Nisha Palackal (Regeneron Pharmaceuticals, Inc. in Tarrytown, NY); Jean-Michel Menet (Sanofi in Vitry-sur-Seine, France); Annette Pieper (Sanofi Industriepark
Höchst in Frankfurt, Germany); and Aurélie Delangle and Annick Gervais (UCB Pharma in Braine L’Alleud, Belgium). The authors also acknowledge the following colleagues for their critical manuscript review: Kudrat Goswami, Peter Salmon, and Gregg Nyberg (Merck & Co., Inc. in Kenilworth, NJ).
References
1 Wang X, Hunter AK, Mozier NM. Host Cell Proteins in Biologics Development: Identification, Quantitation and Risk Assessment. Biotechnol. Bioeng. 103(3) 2009: 446–458.
5 Zhu-Shimoni J, et al. Host Cell Protein Testing By ELISAs and the Use of Orthogonal Methods. Biotechnol. Bioeng. 111(12) 2014: 2367–2379.
7 de Zafra CL, et al. Host Cell Proteins in Biotechnology-Derived Products: A Risk Assessment Framework. Biotechnol. Bioeng. 112(11) 2015: 2284–2291.
8 Chiu J, et al. Knockout of a Difficult-toRemove CHO Host Cell Protein, Lipoprotein Lipase, for Improved Polysorbate Stability in Monoclonal Antibody Formulations. Biotechnol. Bioeng. 114(5) 2017: 1006–1015.
10 Eaton LC. Host Cell Contaminant Protein Assay Development for Recombinant Biopharmaceuticals. J. Chromatogr. A 705(1) 1995: 105–114.
11 Hogwood CE, Bracewell DG, Smales CM. Measurement and Control of Host Cell Proteins (HCPs) in CHO Cell Bioprocesses. Curr. Opin. Biotechnol. 30, December 2014: 153-60.
15 Valente KN, Lenhoff AM, Lee KH. Expression of Difficult-to-Remove Host Cell Protein Impurities During Extended Chinese Hamster Ovary Cell Culture and Their Impact on Continuous Bioprocessing. Biotechnol. Bioeng. 112(6) 2015: 1232–1242.
20 Hall T, et al. Polysorbates 20 and 80 Degradation By Group XV Lysosomal Phospholipase A2 Isomer X1 in Monoclonal Antibody Formulations. J. Pharm. Sci. 105(5) 2016: 1633–1642.
22 Robert F, et al. Degradation of an Fc-Fusion Recombinant Protein By Host Cell Proteases: Identification of a CHO Cathepsin D Protease. Biotechnol. Bioeng. 104(6) 2009: 1132–1141.
23 ICH Q9: Quality Risk Management. US Fed. Reg. 71(106) 2006: 32105–32106; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
24 Dixit N, et al. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 105(5) 2016: 1657–1666.
27 Vanderlaan M, et al. Hamster Phospholipase B-Like 2 (PLBL2), a Host Cell Protein Impurity in CHO-Derived Therapeutic Monoclonal Antibodies. BioProcess Int. 13(4) 2015: 18–29.
28 Gao SX, et al. Fragmentation of a Highly Purified Monoclonal Antibody Attributed to Residual CHO Cell Protease Activity. Biotechnol. Bioeng. 108(4) 2011: 977–982.
29 Labrenz SR. Ester Hydrolysis of Polysorbate 80 in MAb Drug Product: Evidence in Support of the Hypothesized Risk After the Observation of Visible Particulate in MAb Formulations. J. Pharm. Sci. 103(8) 2014: 2268–2277.
30 Lu X, et al. Identification and Quantification of Heat-Shock Protein 70: A Major Host-Cell Protein Contaminant from HEK Host Cells. BioProcess Int. 13(9) 2015: 46–58.
31 Tait AS, et al. Host Cell Protein Dynamics in the Supernatant of a MAb Producing CHO Cell Line. Biotechnol. Bioeng. 109(4) 2012: 971–982.
32 Hogwood CEM, Bracewell DG, Smales CM. Host Cell Protein Dynamics in Recombinant CHO cells. Bioengineered 4(5) 2013: 288–291.
33 Nogal B, Chiba K, Emery JC. Select Host Cell Proteins Coelute with Monoclonal Antibodies in Protein A Chromatography. Biotechnol. Progr. 28(2) 2012: 454–458.
34 Pezzini J, et al. Antibody Capture By Mixed-Mode Chromatography: A Comprehensive Study from Determination of Optimal Purification Conditions to Identification of Contaminating Host Cell Proteins. J. Chromatogr. A 1218(45) 2011: 8197–8208.
35 Sisodiya VN, et al. Studying Host Cell Protein Interactions with Monoclonal Antibodies Using High Throughput Protein A Chromatography. Biotechnol. J. 7(10) 2012: 1233–1241.
36 Shukla AA, Hinckley P. Host Cell Protein Clearance During Protein A Chromatography: Development of an Improved Column Wash Step. Biotechnol. Progr. 24(5) 2008: 1115–1121.
Fengqiang Wang, Daisy Richardson, and Hans-Martin Mueller are with Merck Research Laboratories, Merck & Co. Inc. (Kenilworth, NJ). Georgeen Gaza-Bulseco is with AbbVie Bioresearch Center (Worcester, MA). Xinrong Liu is with ImmunoGen, Inc. (Waltham, MA), as was Fang Liu at the time of this work. Yiwei Zhao is with Takeda Pharmaceuticals (Cambridge, MA). Satish Sharma is with Bristol-Myers Squibb (Devens, MA). Markus Haindl is with Roche Diagnostics GmbH (Penzberg, Germany). Carl Co is with Biogen (Cambridge, MA). Direct correspondence to Tony Gill (gilla@biophorum.com), BioPhorum Development Group (BPDG) facilitator. Since its inception in 2004, the BioPhorum has become a trusted environment in which senior leaders of the biopharmaceutical industry come together to share and discuss openly the emerging trends and challenges facing their industry. BioPhorum currently comprises more than 2,000 active participants in six “phora” covering drug substance, development, fill–finish, a technology roadmap, information technology, and supply partners. The HCP Collaboration is part of the Development Group. This article is a composite view of opinions shared by the whole of the BPDG-HCP and should not be attributed to the individual positions of the participating companies.