Figure 1: Schematic of the thermal core of a differential scanning calorimetry (DSC) system shows reference and sample cells.
The primary goal of biopharmaceutical process development is to determine what steps and conditions will maximize and optimize yields of purified product in the most reproducible, robust, and cost-efficient way. Characterized by high batch-to-batch comparability minimizing economic losses associated with batch failures, success relies on a thorough understanding of a given biological drug. Determining how its activity and stability are affected by processing and how to mitigate and control associated risks is advocated by a quality by design (QbD) approach. However, the complexity and marginal stability of proteins complicates such determinations.
Forward-thinking companies continuously reevaluate the analytical techniques they apply and appraise new technologies to assess their potential value in helping to maintain a business lead. One such technique currently attracting attention is differential scanning calorimetry (DSC). It is a highly discriminating technology for assessing biopharmaceutical stability, already established as the gold-standard method in preformulation development. Generated data are used primarily to inform decisions about critical aspects of liquid formulations: namely, which additives (excipients) and preservatives to use and what buffer characteristics and pH levels will be ideal. Recent innovations have enhanced the practicality of applying DSC further downstream — in process development and manufacturing — to support identification of optimal processing conditions cost effectively.
Here, I review how DSC works, the type of information it generates, and the value it has to offer. Case-study data show how DSC can be used to identify optimal elution conditions during protein A capture step development.
Stability Issues and Process Challenges
The complex structural characteristics of protein-based drug molecules give them much-valued and highly specific activity while substantially increasing the difficulty of their routine manufacture. The primary structure of a protein (its linear sequencing of amino acids) typically is determined and confirmed early in drug discovery. But secondary, tertiary, and quaternary higher order structure (HOS) are studied throughout the drug development process to gain the understanding required to exert effective control and deliver safe and efficacious products.
In solution, a protein exists in a state of equilibrium between native (folded) and denatured (unfolded) conformations. Typically, the correctly folded state is associated with desirable drug activity, making any instability (transition away from the native state) undesirable. The most commonly seen mechanisms of instability fall into one of two categories: biochemical (including oxidation, deamination, and isomerization) and structural (unfolding and aggregation). All these mechanisms change a protein’s HOS, which can adversely affect its clinical performance, and increase immunogenic risk to patients. A drug molecule entering process development is associated with significant economic investment. Thus, the emphasis at early stages is on learning how best to treat an identified candidate so as to maintain its preferred, beneficial HOS through to the point of delivery.
Changes in HOS may be triggered by a broad range of stimuli: e.g., changes in pH and temperature, agitation, shear stress, and light-/chemical-based oxidation. Exposure to such stresses can come at a number of different stages: in manufacturing, storage, shipping, and/or administration. Investigations required for developers to achieve an understanding of the mechanisms of instability, learn how to exert control, and secure a robust operating window can be extensive. Orthogonal techniques are applied to maximize insight, but their effective use relies on understanding how to interpret their multiple datasets correctly.
An Introduction to DSC
The transition of a protein from its native, biologically active state (N) to its unstructured, denatured, and inactive conformation (D) is an endothermic reaction. DSC measures the heat absorbed by a sample as proteins unfold — as a function of temperature — to characterize the unfolding process and consequently the stability of the protein. Figure 1 shows key components of the thermal core of a DSC system.
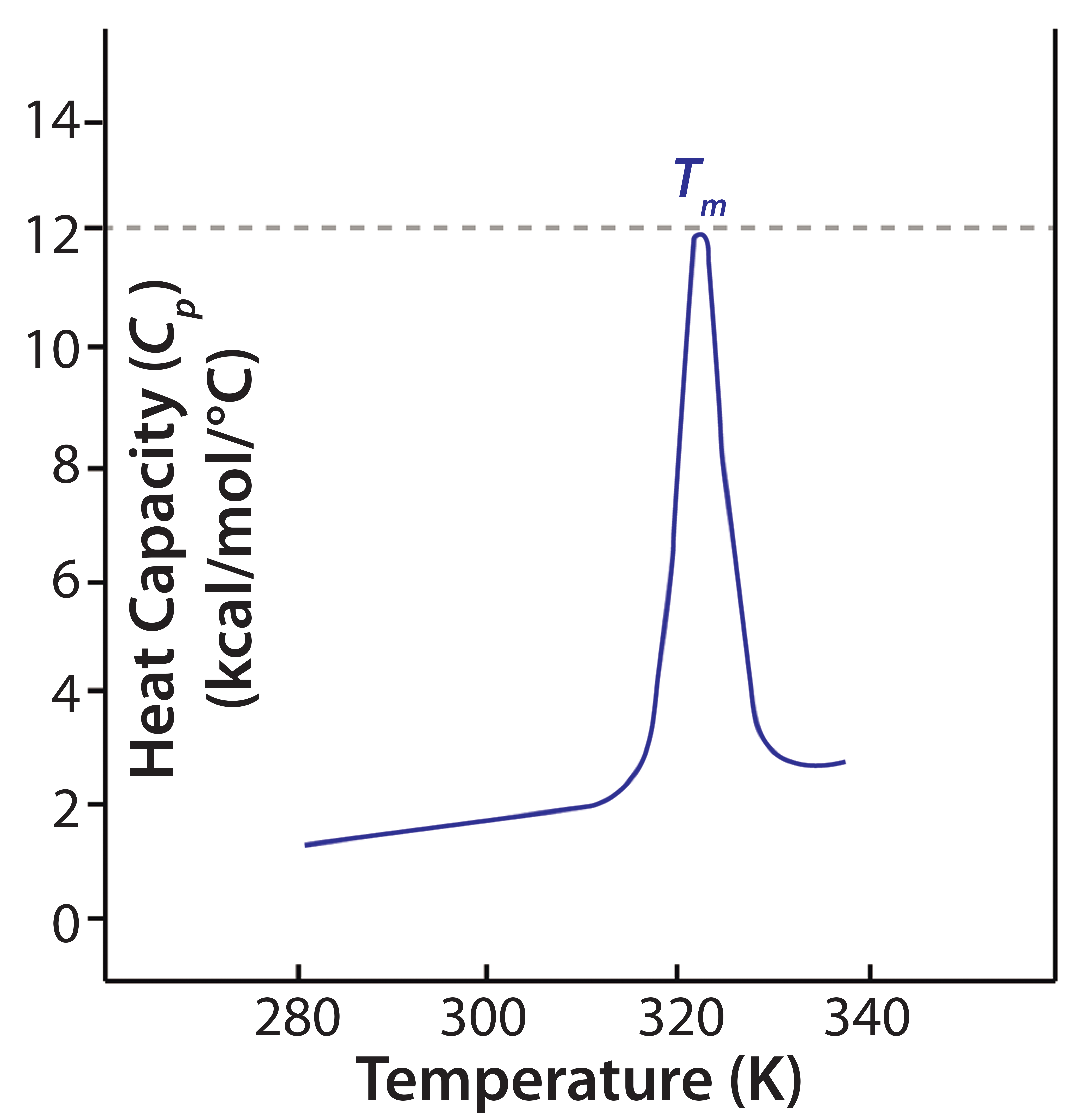
Figure 2: A DSC thermogram shows the denaturation of a single-domain protein; Tm = melting temperature.
During a DSC measurement, both reference and sample cells are heated at a constant rate to maintain constant temperature. Heat is absorbed as protein in the sample cell unfolds and noncovalent bonds are broken, so more energy is required to maintain the cell temperature relative to that of the reference cell. Enthalpy (ΔH) of the unfolding process is calculated directly from that increased energy input. Figure 2 shows a typical DSC thermogram, with enthalpy expressed in terms of specific heat capacity.
The primary metric generated by DSC measurements is Tm: The thermal transition midpoint, melting transition midpoint, or simply the melting point of a protein, Tm is the temperature at which the native and denatured protein are present in equal amounts in a sample. A higher Tm thus indicates higher stability. In practical terms, Tm is the temperature at which a thermogram exhibits its peak value, so it can be determined easily from DSC data. Other information can be determined from a DSC thermogram: the temperature at which unfolding starts (often referred to as the onset temperature) and the enthalpy of denaturation (defined by the area “under the curve”). Large multidomain proteins such as monoclonal antibodies (MAbs) have two to four domains identified in DSC thermograms, each with its own Tm (Figure 4 shows an example).
Together, those metrics enable sensitive comparison of different samples with respect to stability. In process development, this capability is valuable for assessing the impact of processing parameters such as changes in pH and ionic strength during protein purification.
In Practice
Case Study — Identifying Optimal Elution Conditions During Protein A Capture Step Development: Protein A affinity chromatography commonly serves as the initial capture step in MAb production. It binds the Fc region of many immunoglobulin molecules and, when optimized for specificity and selectivity, can yield nearly pure product in a single unit operation. Binding occurs at a neutral pH (around pH 7.0), and elution is at low pH (around pH 3.5). Eluted protein then is neutralized in a high pH buffering system or by a desalting procedure.
A concern with protein A affinity chromatography is that many proteins are unstable at the low pH required for elution. That can cause precipitation, the extent of which usually depends on protein concentration, during or after elution. The net result is that, despite most protein A media being able to bind at least 20 g of antibody per liter of medium, the stability of the protein at low pH becomes a limiting factor. To improve loading capacities and process economics, proteins need to be stabilized in an elution buffer. FUJIFILM Diosynth Biotechnologies scientists investigated an antibody (“protein X”), using DSC to characterize its basic stability at a range of pH values as well as investigating different buffers and additives in an effort to improve process efficiency.
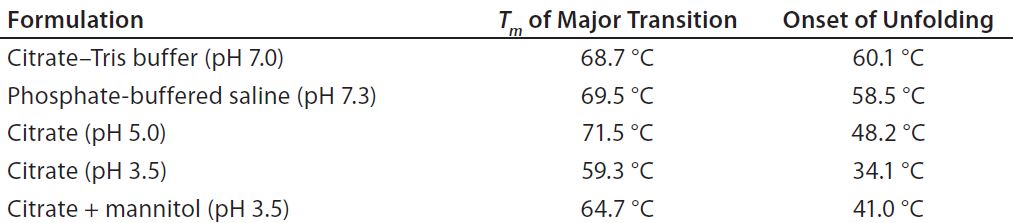
Table 1: Melting temperature (Tm) and unfolding onset temperature of protein X in different buffers as measured by DSC
Preliminary studies indicated that the binding capacity initially was limited to 2 g/L by precipitation of the antibody during elution at higher loadings. To understand the effect of pH on protein X stability, investigators prepared the protein in four different buffers: citrate adjusted to pH 7.0, phosphate at pH 7.3, citrate at pH 3.5, and citrate at pH 5.0. From DSC data (Table 1), protein X showed a higher Tm at pH 7.0 and pH 7.3 than at an acidic pH 3.5, indicating that it was more stable at the higher pH values. Unfolding onset temperatures also registered higher at those more neutral pH conditions. At pH 5.0, protein X had a similar (although slightly lower) Tm to that measured at pH 7, but the unfolding onset temperature was lower. That suggested that pH plays a major role in determining the unfolding behavior of this particular antibody.
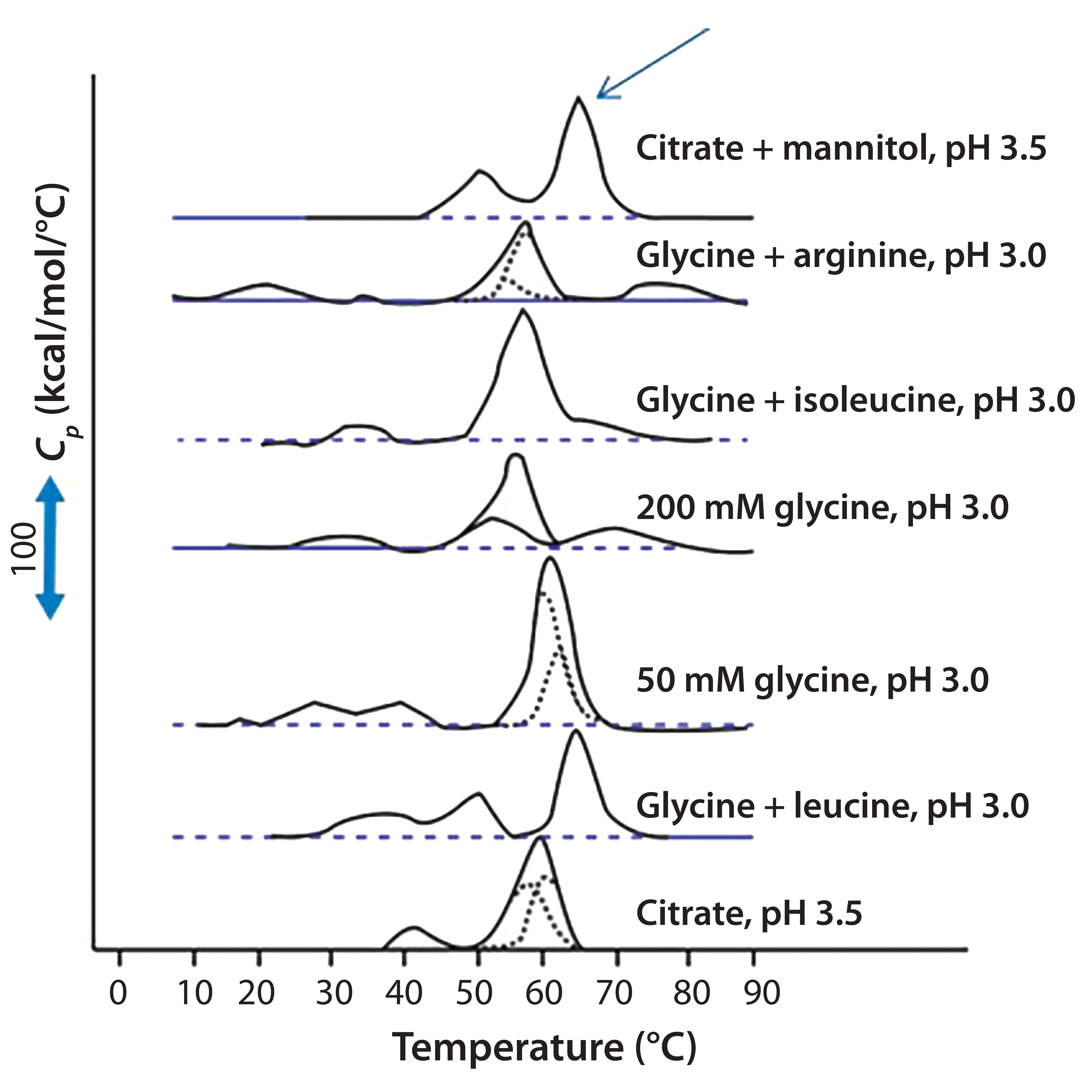
Figure 3: DSC for protein X in seven different elution buffers; black solid lines represent DSC data normalized for protein concentration; broken blue lines represent deconvolution of DSC thermogram data to individual unfolding domains; the arrow indicates buffer conditions that produce the highest Tm (citrate + mannitol at pH 3.5).
Antibody X also was prepared in low-pH buffers with different additives to determine whether any of them could stabilize the protein at low pH levels. DSC data were collected to identify which conditions would increase Tm (Figure 3). Clearly, a citrate buffer with mannitol at pH 3.5 resulted in the largest positive shift in protein X Tm. That indicates the most favorable stability. Adding mannitol increased both Tm and the unfolding onset temperature relative to citrate alone at the same pH (Figure 4).
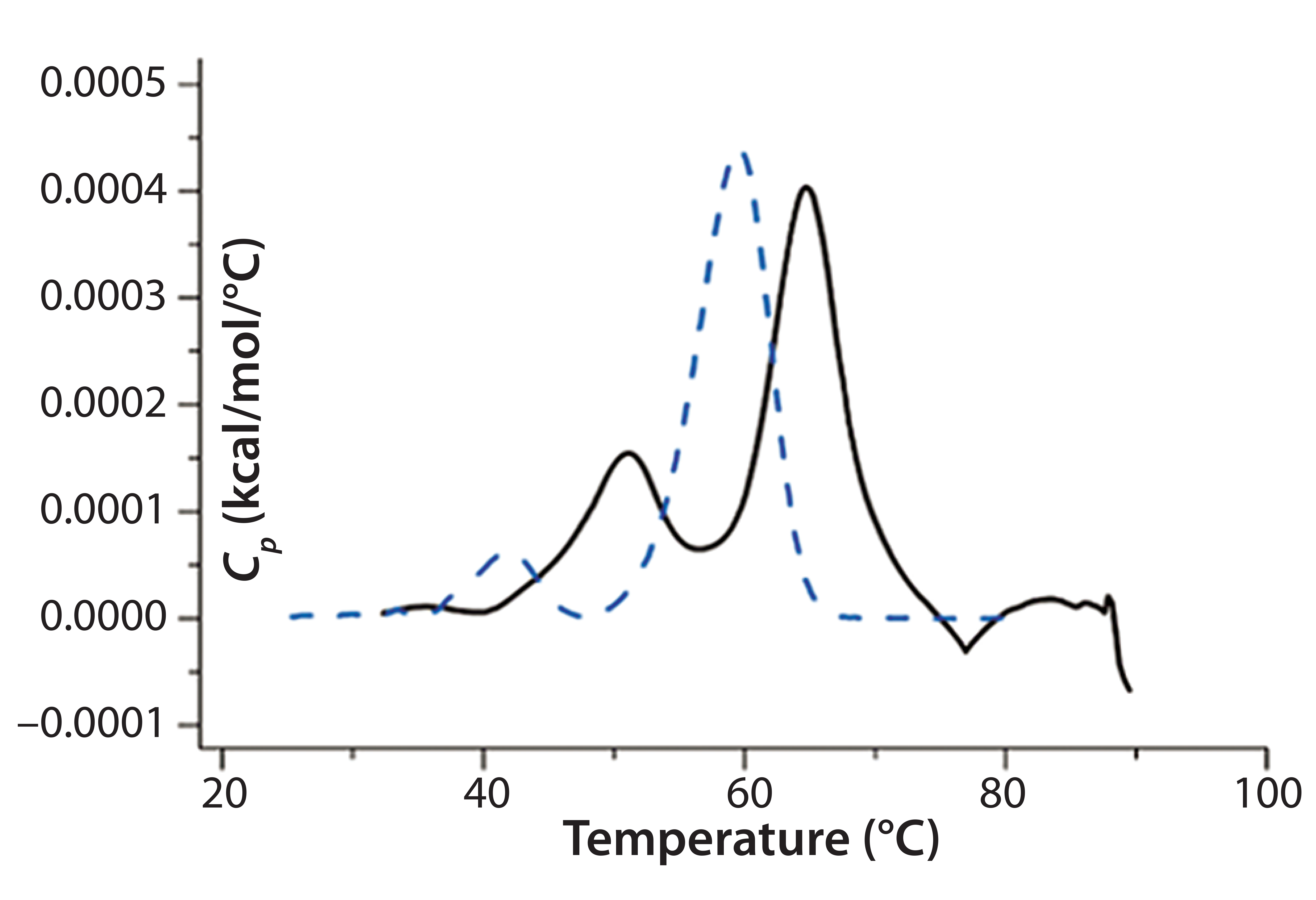
Figure 4: DSC comparing protein X in citrate buffer and citrate buffer + mannitol; the broken blue line represents a thermogram of the protein in the presence of citrate alone, and the black solid line represents a thermogram of the protein in the presence of citrate and mannitol.
In this case study, process development using data generated by DSC resulted in the ability to significantly increase antibody loading during the protein A capture stage. Use of citrate with mannitol as the elution buffer gave at least a 7.5-fold increase in binding capacity to ~15 g/L compared with 2 g/L with only citrate buffer. It is important to note that protein X was more concentrated after the capture step as well, eliminating the need for an ultrafiltration step, which saves materials and processing time (1).
Speeding Development with Powerful Information
DSC data provide a solid basis for comparing formulations during process development. Moreover, using DSC can save time and money once that work advances. Choosing protein-buffering conditions before chromatographic development is undertaken, for example, can provide substantial future savings. New developments in the latest DSC instruments — such as self-cleaning and automated calibration routines and the ability to perform analysis on therapeutic concentrations of protein — build further confidence in the technique. It enables scientists to gather critical data and thus speed up their process development projects.
| New-Generation Solutions for Process Development |
| Much has been written about the desirable characteristics of a technique ideal for stability studies — whether in early stage formulation or process development — and differential scanning calorimetry (DSC) has some inherent attractions in this regard. It is a direct, label-free method with high-throughput capabilities, good reproducibility, and virtually unlimited buffer compatibility. Most important, it can work with samples at typical biotherapeutic concentrations. Furthermore, the data produced correlate well with long-term stability studies, making the technique highly relevant and widely applicable. |
| However, users should factor in a broad range of practical considerations as they transition their techniques toward manufacturing. “Rapid,” “robust,” “easy-to-use,” “cost-effective,” and “automation-ready” appear on most wish lists, but equally important for many organizations is the package of regulatory support available from an instrument vendor. After all, the many guidelines and prescribed methods/procedures present biopharmaceutical companies with a challenge not only to collect data that informs their own decisions regarding drug formulation and manufacturing, but also to ensure that every aspect of analysis meets industry norms and will stand up to regulatory scrutiny. |
| It is important to assess new instruments against all those needs. So how does “new-generation” DSC measure up as a tool for process development? The latest system to come to market (Malvern’s MicroCal PEAQ-DSC) has been designed from the ground up specifically for the biopharmaceutical industry. It comes with full documentation and software support for analytical equipment qualification in line with US Pharmacopeial (USP) and Food and Drug Administration (FDA) guidance, as well as a comprehensive package to support technical compliance with 21 CFR Part 11 and EU GMP Annex 11. In addition, the system is network ready and has built-in automated cleaning and self-validation protocols that minimize downtime to ensure that data reported are of the highest quality. Available dedicated instrument standards help users validate in-assay performance throughout a series of runs. The system can operate 24 hours/day unattended using streamlined workflows and automated data analysis that produce results in hours. |
Reference
1 Acharya P. Application Note AN140623: Manufacturing Biotherapeutic Faster Using Differential Scanning Calorimetry (DSC). Malvern Instruments: Malvern, UK, 2017; https://www.malvern.com/en/support/resource-center/application-notes/AN140623-dsc-for-rapid-efficient-development-of-biopharmaceutical-processes.html.
Verna Frasca is MicroCal product marketing manager at Malvern Instruments Ltd., Enigma Business Park, Grovewood Road, Malvern, Worcestershire, WR14 1XZ, United Kingdom; 44-1684-892456, fax 44-1684-892789; www.malvern.com.