
ADOBE STOCK (HTTPS://STOCK.ADOBE.COM)
Analytical method transfers are essential components of the current global biotechnology environment. Analytical method transfer can be defined as “a documented process that qualifies a laboratory (the receiving laboratory) to use a validated analytical test procedure that originated in another laboratory (sending laboratory), thus ensuring that the receiving laboratory has the procedural knowledge and ability to perform the transferred analytical procedure as intended” (1). The goal is to ensure that a method continues to perform in the validated state regardless of a change in the testing laboratory. Transfers can occur between laboratories at the same site, between sites at the same company, or between companies and/or regulatory laboratories. The requirements for such transfers change throughout the lifecycle of a method.
That background set the stage for the CASSS CMC Strategy Forum “Methods on the Move: Addressing Method Transfer Challenges for the Biopharmaceutical Industry” held on 23 January 2017 in Washington, DC. This forum addressed the current state and challenges of analytical method transfers for late-stage clinical and commercial biotechnology products, including:
- What is the regulatory perspective on method transfers?
- What are the considerations and best practices for method transfers?
- What can we learn from case studies?
Problem Statement
Despite their ubiquitous nature in biotechnology, and unlike some other good manufacturing practice (GMP) activities such as method validation, no definitive regulatory guidelines exist that explicitly define required elements for phase-appropriate analytical method transfer (1–5). The CMC Strategy Forum and this report of it focus on transfer of validated, noncompendial methods (unless otherwise noted).
| CMC Forum Series |
| The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The Forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS (formerly the California Separation Science Society), and is supported by the US Food and Drug Administration (FDA). |
Regulatory Perspectives
Chaired by Sarah Demmon (Eli Lilly and Company), Julia O’Neill (Tunnell Consulting, Inc.), and Timothy Schofield (GlaxoSmithKline), the morning session was titled “Regulatory Compliance.”
Ramesh Potla (CDER, FDA) presented “Regulatory Perspectives on Analytical Method Transfers for Biopharmaceutical Products,” by first defining analytical method transfer and framing transfers in the context of a process validation lifecycle. In this framework, analytical method transfer is analogous to continued process verification, in which assurance is maintained that a method stays in a state of control during routine use.
Potla introduced the discussion of analytical method transfer in the context of risk assessment, then presented several case studies. In one case, transfers of multiple stability-indicating methods were found to be unacceptable because they did not include appropriate aged or spiked samples, resulting in withdrawal of the application. In a second case, attempted transfer of a host-cell DNA method was performed using DNA from a different cell line, resulting in removal of the new site from the application. A third case had no direct comparison between data generated at a receiving laboratory and data for a sending laboratory, and the sponsor did not provide sufficient evidence to support the transfer. In a final case, transfer failed the criterion for assay precision. The sponsor subsequently was asked to perform a full validation of the method at the receiving site and to demonstrate that the newly validated method provided comparable results to those from the originating site. During the panel discussion, it was noted that when a separate method is validated at a second site, the different method validations need to be reconciled. If results of the two validations show meaningful differences, they could indict the current method used to release product.
Hugo Hamel (Health Canada) presented “Regulatory Expectations for Method Transfers: Health Canada’s Perspective.” He provided an overview of Health Canada’s expectations as detailed in the Post-Notice of Compliance (NOC) Changes: Quality Document (specifically Appendix 3 for biologics) (6). This guidance provides detailed information about the types of changes, ways for sponsors to assess them in a risk-based approach, and filing requirements for each type. In general, an analytical method transfer for noncompendial methods requires either protocol preapproval or approval of the method transfer before implementation.
Hamel highlighted three case studies in which issues were observed. In one case, a sponsor attempted to demonstrate suitability of a second laboratory for a potency assay on a new strength of its product using data for the older strength. But a systematic difference was observed with the new strength. Consequently, in the absence of adequate comparative testing, the sponsor was asked to withdraw its application. In a second case, a lack of detail describing sample preparation in a method transfer protocol resulted in differences in protein concentration measured in the transfer and thus failure of the acceptance criteria. In the third case, sponsors relied on specifications for demonstrating comparability between laboratories, which resulted in criteria that were too broadly defined. Hamel stressed that suitable acceptance criteria with the appropriate statistical analyses constitute critical elements of method transfer.
Alfred Del Grosso (CBER, FDA) presented “Analytical Methods Transfer: Considerations for Biological Products.” He focused on framing analytical method transfer in the context of risk assessment and on elements to be considered in risk assessment. Del Grosso detailed a list of analytical transfer issues to consider, including instrumentation differences, training and analyst experience differences, reagent differences, and software differences. He also mentioned unrealistically tight or overly broad acceptance criteria, insufficient number of samples or data points, failure to identify bias between laboratories, and minor differences in the execution of a test method between laboratories (such as minor equipment, sample preparation, and calculations).
Industry Perspectives and Best Practices
Discussion of industry perspectives, best practices, and case studies continued in the afternoon session, chaired by Mark DiMartino (Amgen), Hamel, and Ned Mozier (Pfizer). This session was titled “Key Ingredients Needed for Successful Method Transfers: Cases Included.”
Dawn Sailer (Eli Lilly and Company) presented “Biopharmaceutical Product Method Transfer Studies Enabling Global Launch: Strategies, Case Studies, and Lessons Learned.” She detailed Lilly’s analytical method transfer strategy, which includes predefined sample requirements that differentiate laboratories intended for release/stability testing from those intended for in-country testing.
For release/stability testing laboratories, the strategy includes four samples (drug substance, stressed drug substance, drug product, and stressed drug product), quantitative acceptance criteria for unstressed samples, and qualitative acceptance criteria for stressed samples.
For in-country testing laboratories, studies include only one drug product sample. Study designs include a statistically based number of setups (4–12) with acceptance criteria for all attributes on specification, including comparison of means between laboratories and variability at a receiving laboratory. For bioassays, training is performed routinely in-person at a receiving laboratory minimally for the first execution of a particular method, and three successful executions are required.
Bioassay transfers are performed using a two-tiered approach: If 10 method executions do not meet acceptance criteria, then 6–10 additional executions are performed and assessed against tighter acceptance criteria. Sailer detailed case studies for compendial method transfers where the root causes of issues were related to a lack of method clarity (e.g., due to translation from English to the language of the receiving laboratory) and method failure posttransfer because of a change in a reagent vendor.
Sailer presented case studies of issues relating to transfer of noncompendial methods. In one case, the investigation revealed a time-dependent increase in measured protein concentration because of a leachate from tubes used in the receiving laboratory. Results highlighted the importance of understanding in totality the equipment and reagents used by a receiving laboratory. In a second case, an atypical peak observed in a capillary electrophoresis sodium dodecyl sulfate (CE-SDS) method was caused by the local temperature at the receiving laboratory, which led to incomplete reduction. That demonstrated the importance of knowing a laboratory’s environmental setup.
In a third case, an international receiving laboratory experienced a technical equipment issue that was commonly known and had been mitigated in systems in the United States. That case study revealed a lack of internal communication at a global vendor. In a case study for a cell-based assay, unexpected cell growth at a receiving laboratory was linked back to inappropriate qualification of an automated cell counter. In another bioassay, unexpected high results at a receiving laboratory were a result of an incorrectly calibrated electronic pipette. An issue observed with an additional cell-based bioassay was found to be caused by an independent cell bank prepared and inappropriately maintained at a receiving laboratory. In a final example, no signal was observed in a cell-based assay because of inappropriate subjection of the cells to trypsin.
Greg Frazer (Pfizer) presented “Case Studies and the Validation and Transfer of Analytical Methods for MAbs and Vaccines to Enable Commercial Launch.” He highlighted Pfizer’s analytical method transfer strategy, which included more than 125 different method validations and transfers to support late-stage biological compounds over the past two years. The transfer strategy emphasizes in-person knowledge sharing and training. Typical validation/transfer timelines are on the order of 12 months, with emphasis on upfront planning, feasibility, and familiarization. The company’s preferred strategy is to include multiple laboratories in a covalidation. If that is impossible, then an additional laboratory can be added to the original validation report using a supplemental protocol.
Frazer detailed the practice of characterization of method performance using total analytical error (TAE), which is a measure of method bias from the true value plus three standard deviations (SDs). Acceptance criteria in analytical method transfers account for the characterized TAE, where the receiving laboratory means must be no more than one-third TAE relative percentage to the transferring laboratory for purity/impurity methods and one-half TAE for assay and process impurity methods. That ties the demonstrated method performance, method validation, and transfer together as part of a successful method transfer. Frazer provided a case study for a cell-based method in which thorough method characterization and transfer preparation resulted in outstanding method performance at both sites with RSDs ≤7% and mean differences <2% for three sample levels.
Nadine Ritter (Global Biotech Experts LLC) presented “Practical Points in Method Tech Transfers: Three Savory Scenarios.” She stressed the complexity of analytical method transfers in the context of diverse molecule and method types common in the biotechnology industry. Based on extensive experience working with clients across the industry, Ritter recommended selecting a transfer strategy based on the similarity of systems between transferring and receiving laboratories. That includes covalidation or an intermediate precision-like transfer study for identical systems between laboratories, repeat of key validation parameters for similar systems between laboratories, and revalidation in cases of adaptation of a method to new systems. She emphasized the importance of a complete verification of the intended use of a method at the new site and the technical differences in the execution of the method at the receiving laboratory. Finally, the new laboratory should track and trend the method performance after transfer.
Jeffrey Staecker (BioPhia Consulting, Inc.) presented “Risk-Based Approach to Method Transfer.” He discussed the process and benefits of risk-based approaches to analytical method transfer. He also presented on the use of failure mode and effects analysis (FMEA) components of severity, probability, and detectability of a failure as a model for application to transfers.
Severity of failure for a particular method and product can be assessed by looking at potential effects on safety and efficacy. A case-by-case analysis of a specific method’s failure to work for a specific product can include items such as how the method fits into the overall control strategy and whether there’s a similar orthogonal method.
Probability of failure can be estimated using data from Cpk analysis (an analysis on process capability) of a bioprocess. A lower Cpk (an index of how close a process is running to its specification limits) indicates that a process is near a specification where a method transfer affecting bias or precision might have a high probability of influencing product disposition. For processes with a good Cpk (e.g., >1.33), a small amount of bias or imprecision introduced from method transfer is unlikely to affect a product. This approach can be used to estimate risk regardless of whether the low Cpk is driven by process or method.
Failure can be detected by appropriate method monitoring (e.g., statistical process control using the same references as in testing by a sending laboratory).
A benefit of a risk-based approach to method transfer is to ensure the appropriate level of work and rigor for a specific transfer exercise while simultaneously reducing risks associated with that transfer.
Julie Frost (Amgen) presented “A Balancing Act: Streamlining Method Transfers Without Compromising Compliance or Science.” She discussed the business-driven need to minimize resources spent on analytical method transfers while ensuring comprehensive demonstration of the suitability at a receiving laboratory, describing Amgen’s strategy to balance those competing priorities. Amgen uses historical data from transferring laboratories when possible. Advantages of doing so include developing a more accurate picture of method performance over time and building a larger available data set. It also allows transferring staff to focus on assessment of method performance in a receiving laboratory. That approach applies best for later stage (mature) products because recently approved products do not have large historical data sets that can be used for comparison.
For quantitative methods, equivalency and precision criteria are set based on historical data and method validation results. Performance at a receiving laboratory is assessed using a statistical test of equivalence based on two one-sided tests (TOST) with a two-sided 90% confidence interval. For qualitative methods, criteria are based on specifications, and results at a receiving laboratory are evaluated without statistical analysis.
Frost presented a study design that would be executed in a receiving laboratory. In this design, equivalency and intermediate precision are assessed using 24 replicates in eight assays for which a reference standard or assay control samples are tested. This design encompasses method-specific sources of variability (e.g., analyst, day, instruments). All applicable sample types (e.g., in-process, stability, or forced degradation samples) are verified under conditions of use. Posttransfer method trending is incorporated into quality systems.
Summary of Learnings and Discussions Throughout the Forum
Strategies for Analytical Method Transfer: Generally, transfers of validated, noncompendial methods can be accomplished using four main strategies: covalidation, comparative testing, revalidation, and transfer waivers.
Covalidation between two or more laboratories can be used to demonstrate that a method is suitable for its intended use at multiple sites. Generally, covalidation is used at the time of biologic license application (BLA)–enabling method validation activities performed in accordance with ICH Q2(R1). Reproducibility between test sites is assessed using predefined acceptance criteria.
Comparative testing is another common way to accomplish analytical method transfer. Here, a method previously demonstrated to be suitable for its intended use at one laboratory (through validation or prior transfer) is transferred to a second laboratory. The transfer activity includes a predefined number of replicates of testing the same sample(s) at both sites for an appropriate subset of the ICH Q2(R1) validation parameters. Predefined acceptance criteria are used to determine whether a method performs similarly at the transferring and receiving laboratories.
Revalidation is a strategy through which a method may be demonstrated suitable for its intended use at a laboratory separate from the laboratory involved in initial method validation. Revalidation may be full or partial based on the degree to which the appropriate ICH Q2(R1) method parameters are reassessed at the new laboratory. There are multiple drivers for revalidation as a method transfer strategy. One common application is when the laboratory at which a method was initially validated is unavailable to perform testing activities in support of method transfer.
Use of a transfer waiver is defined in USP <1224>. However, the FDA has stated that transfer waivers are not appropriate for transfer of validated, noncompendial methods for biotechnology products.
Each method and product should be assessed as a unique situation. Careful consideration of the appropriate strategy to accomplish method transfer is critical to minimize resource use while ensuring robust method transfer and facilitating regulatory approvals.
Criticality of Project Management and Planning: Strong project management is essential to setting up a successful analytical method transfer. The need for project management increases with complexities such as tight timelines, time zone or language differences, scope (including multiple and/or complex methods), and specific regional requirements. A typical method transfer timeline can be from six months to a year or longer depending on complexity. Investment of resources in high-quality project management, upfront planning, and relationship building often are areas where corners might be cut when resources or timelines are tight. However, they are the fundamental nontechnical aspects of an analytical method transfer project that will determine its success or failure and could reduce time on the back end of a transfer (because of misalignment, miscommunication, or investigations). Face-to-face interactions between transferring and receiving laboratories are invaluable to build team relationships and define and understand the intangibles that can affect method transfer.
Risk Assessment As a Driver for Transfer Study Design: As outlined in ICH Q9, a risk-based approach is a powerful tool to design analytical method transfer studies, determine what data are required, and define what success will look like (7). Assessment should include a combination of the severity, probability, and detectability of known risks associated with a transfer, with considerations including but not limited to
- Method type (qualitative or quantitative), intended use, and method historical performance (consistency, robustness)
- Laboratory experience with product (manufacturing, testing history, Cpk, total analytical error)
- Method criticality with respect to critical quality attributes (CQAs)
- Procedural complexity
- Alignment of test method across sites
- Sample types and availability for testing samples from same product lots by both laboratories
- Requirements for statistical analysis.
An industry example of risk-based analytical method transfer is an evaluation of risks during robustness studies as high, medium, or low. Such an evaluation also includes high or medium risks in transfer studies. Regulator feedback on the application of risk assessment for analytical method transfer was overwhelmingly positive. Health Canada representatives indicated that risk assessment is not included in most submissions, but that failure mode effects analysis (FMEA) is the most commonly observed tool for method transfers when risk assessment is included in submissions. FDA representatives stated that the agency would like to see the scope of the differences and changes between laboratories defined in a submission so that an accurate independent assessment can be made. Risk should be defined in the scope of such changes.
Gap assessment for evaluation of all method-related information (including development and validation reports and method procedure) should be performed well in advance of initiating the activity. Site visits are extremely valuable at this stage. Alignment of a method and available equipment and materials between transferring and receiving laboratories is critical to evaluate as a part of a study design. Considerations include instruments, equipment, software, peripheral equipment, reagents, standards, and shipping/handling. Local characteristics specific to the transferring or receiving laboratories (e.g., voltage, elevation, humidity, ambient temperature, and solar radiation) can effect significantly the ability to execute the method, so such factors must be considered. Often overlooked, sample handling frequently is a significant source of variability and thus should be a primary area of focus during information sharing and training. Another critical factor to consider is how data integrity will be ensured and maintained, both during method transfer and posttransfer during routine testing.
Both laboratories should participate in developing a method transfer protocol. It should include clearly defined roles and responsibilities, the study design, samples and reference standards to be used, acceptance criteria, deviation policy and reporting, oversight, and approval authority.
Sample selection is an essential component of analytical method transfer and should be considered relative to the method type and intended use at a receiving laboratory. Samples should be designed to address risks identified in the scope of an analytical method transfer exercise. Appropriately, stressed or forced-degraded samples can be used to challenge a method’s limits. Regulators have not provided general guidance on the number of lots required. Historical expectations may have been to include three samples (e.g., from three lots of material or three samples prepared from a single lot), but fewer samples can be acceptable if justified appropriately. For example, if intended use of a method to be transferred is for testing both drug substance and drug product, then both sample types should be included in that transfer unless it is justified to be unnecessary (e.g., both samples have the same formulation). The FDA and Health Canada indicated that a single lot of material would be acceptable if justified — which can be addressed when assessing the need for additional specificity information as part of a transfer.
Industry representatives provided examples for sample-selection options. Use of validation samples for transfer is a best practice and streamlines transfer because data for a transferring laboratory may be available already. If suitably justified, a comparative transfer study with a single sample such as a reference standard or assay control can be coupled with a verification exercise for other sample types.
During a panel discussion, it was mentioned that use of multiple lots of material can introduce process variability into transfer data, which can make it more complicated to understand changes because of method transfer. For cases in which multiple lots of material are required for method transfer studies, the study design and analysis must incorporate proper statistical treatment of the data. Otherwise, incorrect conclusions might be drawn. In general, industry representatives saw less value in using multiple samples for method transfer and questioned whether other studies might not be more appropriate if specificity concerns related to method transfer arose. When statistical methods are used for method transfers, more attention should be given to replications of a method than to the number of samples. Likewise, the number of manufactured lots at a similar level is less important than samples that span a range in an assay.
Analysts, Equipment, and Training: Selection of the number of analysts and their training requirements should take into consideration the “risk level” of analysts to study results as well as analysts’ skill sets, level of training, and history with similar methods. General guidance is to include a minimum of two analysts at a receiving laboratory because more than one analyst is required to compare intermediate precision at the two laboratories. Because the intent of an analytical method transfer is to demonstrate suitability of a laboratory and not specific analysts, a receiving laboratory must have suitable analyst training and qualification procedures and a plan to maintain those levels after completion of a method transfer. It is also important to assess equipment qualification at a receiving laboratory and not to assume that vendor knowledge and support are equivalent globally.
Information sharing and training between transferring and receiving laboratories is critical for success. Documentation sharing (such as standard operating procedures, validation reports, certificates of analysis for standards and controls, and sample-handling procedures) is a minimum requirement. In-person training always is a best practice to increase the likelihood of success. For simpler methods or those with which a receiving laboratory has significant experience, e-learnings such as training videos or teleconference-based trainings have proven to be viable strategies.
Setting Acceptance Criteria: Successful analytical method transfer is based on a robust method with high-quality validation. Acceptance criteria are critical components for an analytical method transfer to meet its intended goal. If criteria are too wide, a real difference between both laboratories might not be detected. If criteria are too narrow, issues might be detected that do not affect practical results for a method.
Many approaches can be used to establish transfer acceptance criteria — ranging from adapting validation acceptance criteria to using Cpk knowledge and adopting a statistical approach after establishing an acceptable level of product risk. Historical data from repeated measurement of the same sample over time (e.g., reference standards, assay controls, and stability samples) may be indicative of method performance. It is important to ensure that a historical data set used as a benchmark includes all sources of normal variability (e.g., multiple instruments, analysts, reagent lots, and column lots). Other approaches include basing requirements on specifications that a laboratory will be testing against or basing acceptance criteria on the total analytical error of a method relative to specification (e.g., using Cpk information).
Results and Performance Monitoring: In addition to a formal statistical comparison of results at transferring and receiving laboratories, rates of invalid assays should be compared. An established baseline invalid assay rate should be based on historical method performance at a transferring laboratory. It is reasonable to expect a similar rate of invalid assays at a receiving laboratory. If a receiving laboratory experiences a meaningful increase in invalid rates, that should be evaluated because it might suggest an issue with a method transfer. Manufacturers should understand such assay failures at a receiving laboratory during transfer because they can be early indicators of an issue to be remediated. Those failures should be investigated in real time before transfer testing activities continue. Speakers from the FDA indicated that the agency has no set expectations on numbers of invalid assays, but an example was provided by one industry speaker in which a non-US agency did expect to see some invalid assays to confirm that system suitability criteria are effective. A zero invalid rate for certain types of assays can reflect an inappropriate validity criterion.
Method monitoring after completion of a transfer is excellent for risk mitigation. Monitoring after transfer also provides ongoing data collection beyond the point-in-time data generation of a method transfer study. Trending the pattern of root causes for invalid assays can differentiate expected sources (e.g., aging columns, bad batches of reagent) from specific causes that should be assessed (e.g., insufficiency of training program or lack of method robustness). Trending the performance of standards can confirm expected precision and early warning of assay drift to prevent or support investigations of unusual release or stability results. Each time a transfer activity occurs, the infrastructure should be ensured for comprehensive long-term method monitoring at all laboratories that will run a method.
Statistical Considerations: As with the setting of acceptance criteria, statistical design and analysis are critical components of a transfer study. If method experience and the available historical data set are limited, it might be necessary to perform a more robust transfer study. Formal statistical methods such as TOST should be considered for transfer of methods that measure attributes related to a product’s safety and efficacy. Less rigorous approaches can be used for methods on attributes that are less related to clinical outcomes.
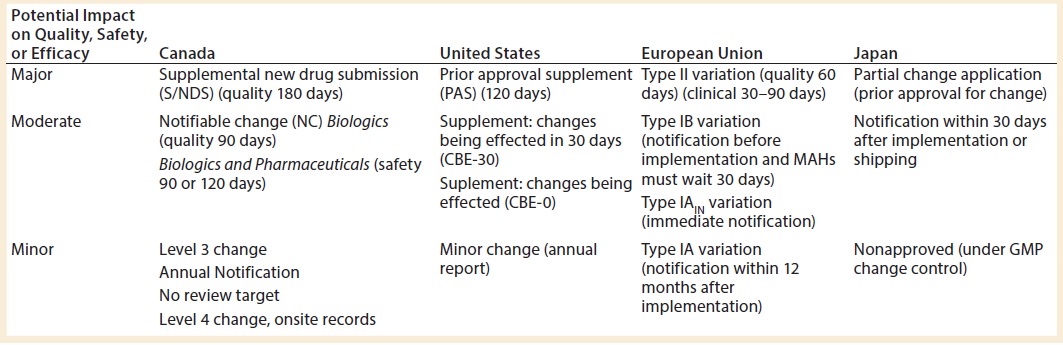
Table 1: Reporting requirements for analytical method transfers
Reporting requirements for analytical method transfers vary with the market and with the assessed impact of change on quality, safety, or efficacy. Hamel presented a table summarizing requirements for some major markets (Table 1). Each situation should be assessed independently in the context of appropriate regulations.
Forum Progress
The biotechnology industry’s growing global complexity and requirements of some markets to perform in-country product release are leading to unprecedented levels of analytical method transfers in the absence of clear harmonized expectations for the scope and depth of transfer activities. The 2017 CMC Strategy Forum “Methods on the Move: Addressing Method Transfer Challenges for the Biopharmaceutical Industry” took steps toward addressing the existing gaps through a collaborative discussion between industry and regulators discussing expectations, strategies, best practices, and case studies.
| CMC Strategy Forum North America Program Committee |
| Siddharth Advant (Celgene Corporation), Yves Aubin (Health Canada), John Bishop (CBER, FDA), Barry Cherney (Amgen Inc.), JR Dobbins (Eli Lilly and Company), Julia Edwards (Allergan), Sarah Kennett (CDER, FDA), Joseph Kutza (MedImmune, a member of the AstraZeneca Group), Kimberly May (Merck & Co., Inc.), Anthony Mire-Sluis (AstraZeneca), Stefanie Pluschkell (Pfizer, Inc.), Nadine Ritter (Global Biotech Experts, LLC), Dieter Schmalzing (Genentech, a member of the Roche Group), Timothy Schofield (GlaxoSmithKline), Zahra Shahrokh (ZDev Consulting), Jeffrey Staecker (BioPhia Consulting, Inc.), Andrew Weiskopf (Biogen), and Marcel Zocher (Bristol-Myers Squibb Company) |
| Disclaimer |
| The content of this manuscript reflects discussions that occurred during the CMC Strategy Forum. This document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance documents, points-to-consider documents, or direct discussions with the agency. |
References
1 USP <1224> Transfer of Analytical Procedures. USP–NF. US Pharmacopeial Convention: Rockville, MD.
2 Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. US Food and Drug Administration: Silver Spring, MD, 2015.
3 ICH Q2(1): Validation of Analytical Procedures. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 1994.
4 Guidance for Industry: PAC-ATLS: Postapproval Changes — Analytical Testing Laboratory Sites. US Food and Drug Administration: Silver Spring, MD, 1998.
5 PDA Technical Report 57: Analytical Method Validation and Transfer for Biotechnology Products. Parenteral Drug Association: Bethesda, MD, 2012.
6 Guidance Document: Post-Notice of Compliance (NOC) Changes: Quality Document. Health Canada: Ottawa, ON, 2016 (revised); www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/postnoc_change_apresac/noc_pn_quality_ac_sa_qualite-final-eng.pdf.
7 ICH Q9: Quality Risk Management. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland.
Mary Beth Pelletier, PhD, is associate director at Biogen. 5000 Davis Drive, Research Triangle Park, NC 27709; marybeth.pelletier@biogen.com; Jeffrey Staecker, PhD, is principal consultant at BioPhia Consulting, Inc.; jstaecker@biophia.com. Kathy Lee, MS, is senior research advisor, global regulatory CMC at Eli Lilly and Company; Lee_kathy2@lilly.com.