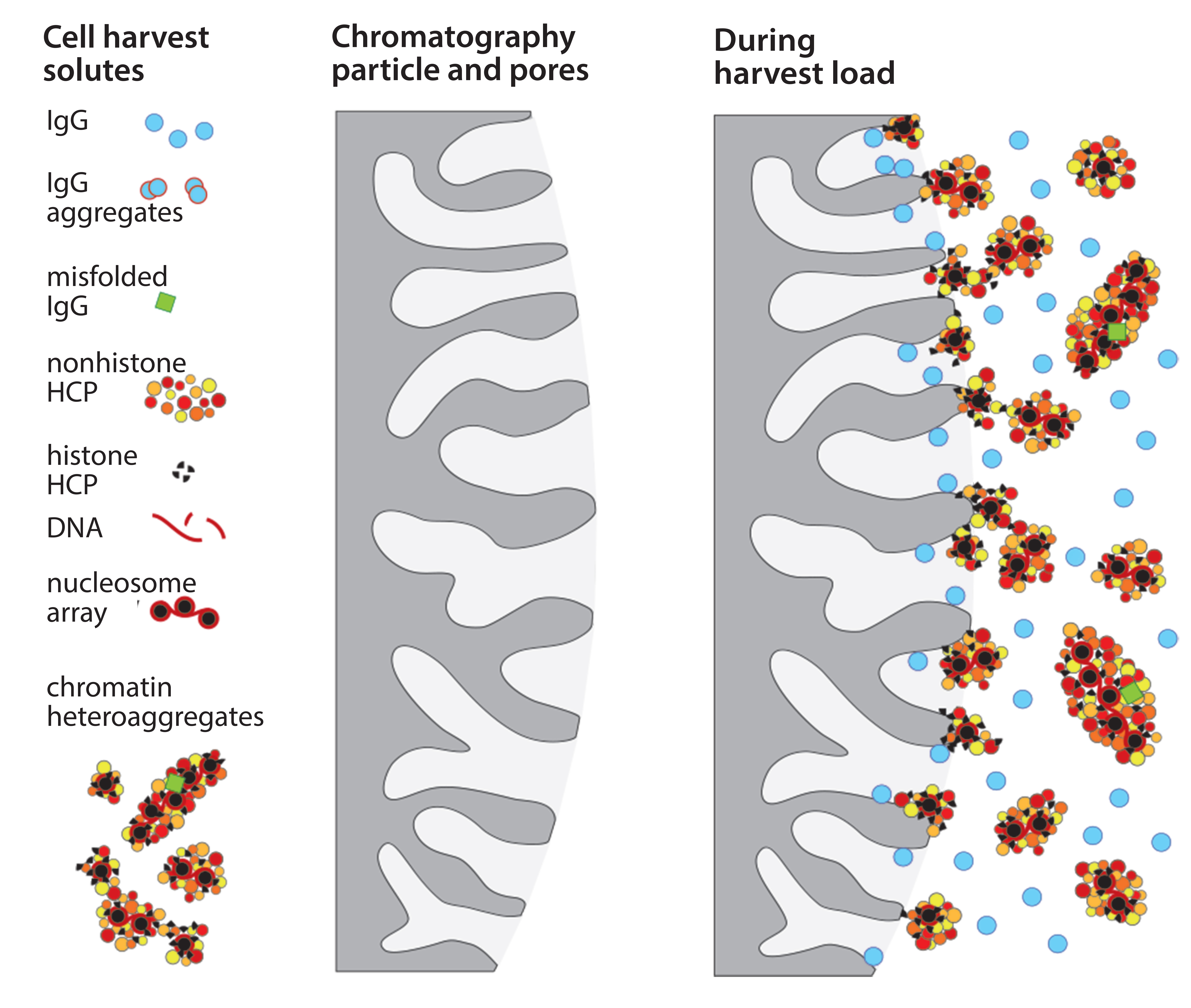
Figure 1: Fouling of chromatography particle surfaces by compound contaminant associations
One of the early disappointments in development of immunoglobulin G (IgG) purification technology was ultrafiltration on membranes with 50–100 kDa cutoffs. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) showed that most host cell proteins were smaller than that. IgG was retained. Parallel concentration and buffer exchange could be performed going into a follow-on polishing step. These features made it an obvious candidate for initial capture, but it did not perform as hoped. Membrane fouling sabotaged its concentration–diafiltration potential, and prohibitive levels of host contaminants remained in the IgG fraction. Protein A affinity chromatography became established as the preferred capture method. Ultrafiltration was consigned to the supporting role of concentration and diafiltration after purification was essentially complete.
Protein A has since proven to be consistent and highly competent, but like ultrafiltration, it should be capable of much better performance than it generally delivers. In its prechromatography days, it was hailed for its exquisite specificity, binding only IgG. The expectation was that the affinity chromatography version would achieve essentially complete purification in a single step. In practice, host cell protein (HCP) contamination often is observed in the range of 1,000–10,000 ppm. DNA also persists. It does not seem reasonable that protein A should have affinity for either, and it doesn’t. Their persistence points instead to a nonspecific contamination pathway.
Depressed protein A performance is now understood to result mainly from chemical fouling of chromatography media during sample application. Chemical fouling is distinct from physical fouling. Physical fouling involves cells and debris of a size sufficient to clog filters and columns. Chemical fouling involves nonspecific chemical interactions of contaminants with the surfaces of purification media; 20–40% of host contaminants in cell culture harvests are associated in compound assemblages that include hundreds of species (1–3). Some are weakly associated, some strongly. Some of their components bind more strongly to protein A than IgG does and act as anchors for associated species that individually lack affinity for the chromatography surface.
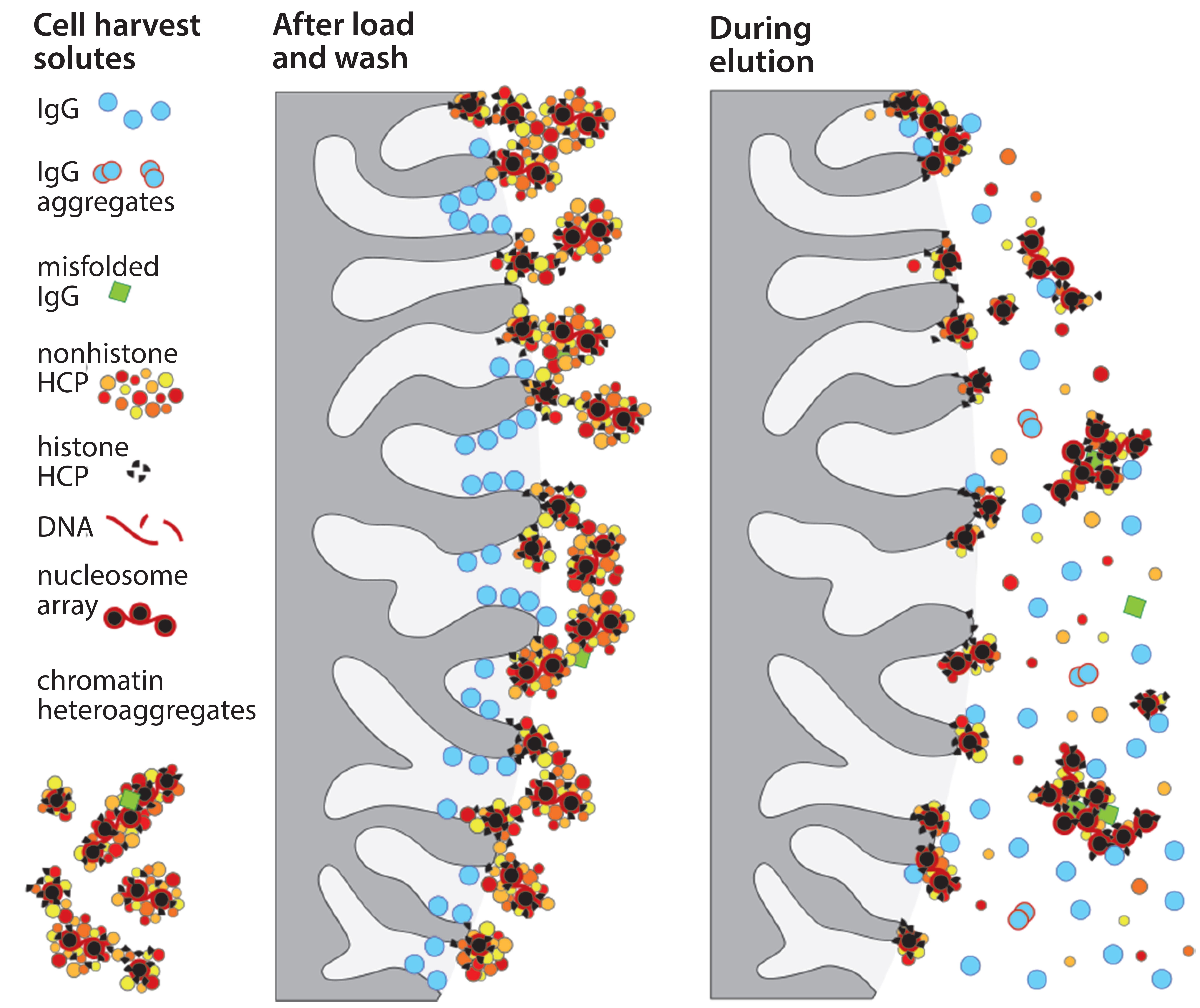
Figure 2: Dissociation of contaminant subsets coincident with IgG elution
Accumulation of such assemblages on chromatography surfaces interferes with performance in two major ways. Aggregates of 50–400 nm block IgG access to the diffusive pores in chromatography particles. That reduces dynamic capacity on protein A as much as 20% compared with loading purified IgG (Figure 1). IgG elution conditions subsequently dissociate contaminant subsets from still-anchored components, and the dissociated subsets cooccupy the IgG fraction (Figure 2). 99% of host contaminants in IgG eluted from protein A derive from this pathway (1).
The effects of chemical fouling are not limited to protein A. They compromise all chromatography methods. They reduce capacity on traditional and multimodal cation exchangers by 50–65% (4, 5). Chemical fouling burdens even size-exclusion chromatography (SEC), in which the chemical surfaces of chromatography particles are generally assumed to be inert. In fact, SEC interacts so strongly with some contaminant heteroassociations that they smear all the way across the elution profile instead of being restricted to the aggregate fractions corresponding to their actual size class (6).
Removing chemical foulants before column loading enables dramatic improvement: HCP removal by protein A improves more than 100-fold and DNA removal more than 1,000-fold. IgG binding capacity is elevated to the same levels obtained when protein A is loaded with purified IgG (3). Improvements with ion-exchange and multimodal capture are even greater (4, 5).
Given that chemical fouling depresses performance of all chromatography methods and that advance removal of chemical foulants enables them to fulfill their fractionation potential, could it also enable ultrafiltration to deliver the potential envisioned in the 1980s? This article addresses that question and goes a step further. It coordinates capture by ultrafiltration with an in-line polishing chromatography step that takes advantage of ultrafiltration’s abilities to perform parallel concentration and diafiltration.
Removing Chemical Foulants from Cell Culture Harvests
Advance removal of chemical foulants from cell culture harvests targets the most reactive species: the most negatively charged, the most positively charged, the most hydrophobic, largest and least soluble. This is accomplished by adding a combination of flocculating agents to cell-free or cell-containing harvests. Allantoin crystals bind aggregates, viruses, and endotoxins by hydrogen bonding (7–9). Octanoic (caprylic) acid precipitates a variety of HCP and viruses (3–5). Electropositive polymers, particles, or depth filters target DNA, virus, and acidic HCP. Removal of solids eliminates 40–70% of HCP, 99% of DNA, 2–3 logs of endotoxin, 5–9 logs of virus, and 75–95% of aggregates (1–5). Typical IgG losses of about 10% mostly represent misfolded product associated with aggregates. Other approaches are discussed in the literature (10–12).
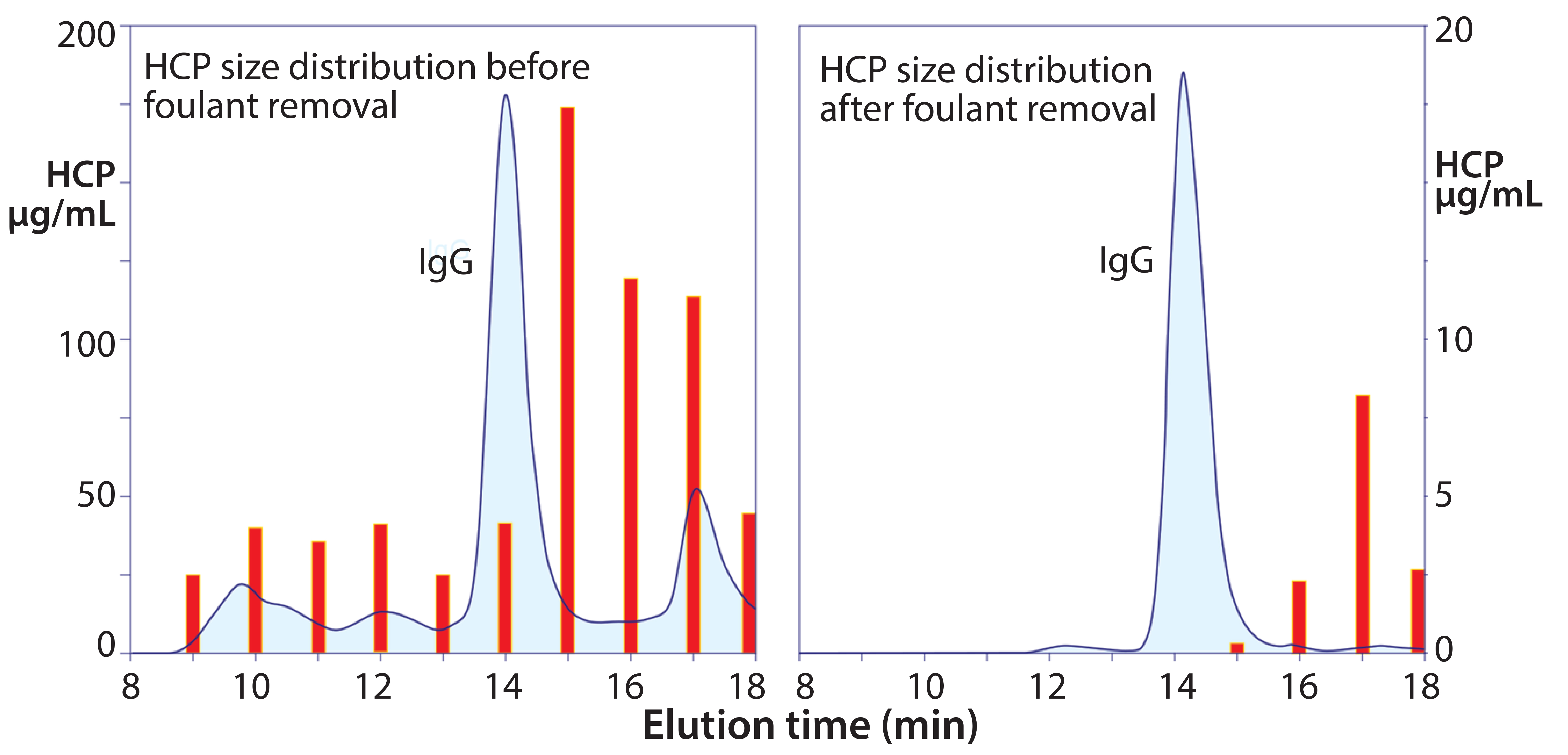
Figure 3: Size distribution of host cell protein (HCP) before and after foulant removal; note the different scales on the left and right frames.
Figure 3 illustrates SEC profiles before and after flocculation of a Chinese hamster ovary (CHO) cell culture harvest. The aggregate population — which has nearly the same cumulative mass as the IgG peak — contains misfolded IgG, but the dominant species are HCP associated with chromatin nucleation centers (1–5). This illustrates why ultrafiltration initially failed to meet expectations as an IgG capture method. The aggregates would have been coretained with the IgG where they interfered with pore flux and remained in the retentate with the IgG after processing. The right-hand frame in Figure 3 shows why advance foulant removal should enable ultrafiltration to deliver outstanding performance.
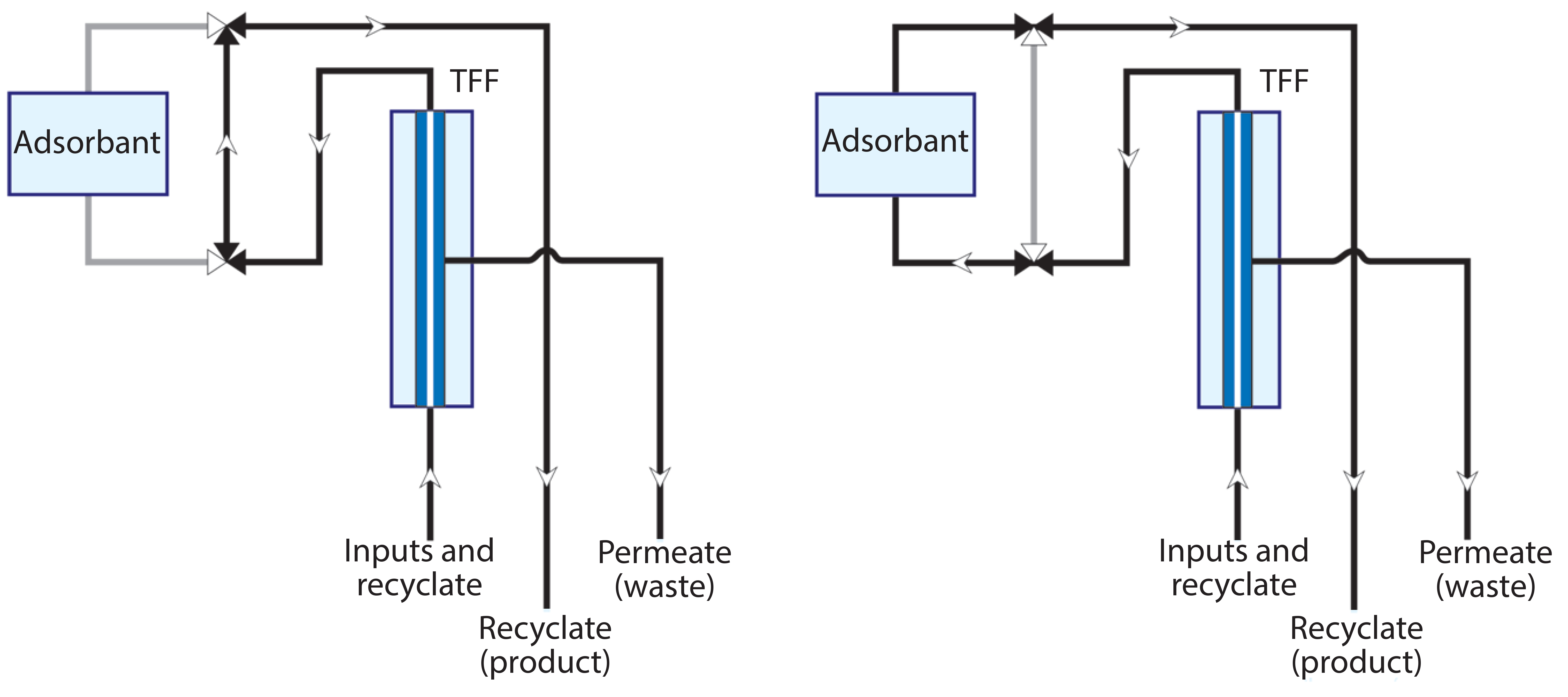
Figure 4: Flow diagrams through an apparatus configured to support ultrafiltration with a single adsorbent channel
Ultrafiltration-Adsorption for Integrated IgG Capture–Polishing
Figure 4 illustrates a basic apparatus for coordinating ultrafiltration with in-line adsorption (13–15). The first frame shows the system with only an ultrafiltration unit in line. The second frame illustrates the flow path with an adsorptive chromatography unit also in line. Additional adsorbent channels can be added, and any given adsorbent can be operated in either bind–elute or flow-through mode. Figure 5 shows an early prototype with two adsorbent channels.

Figure 5: An early laboratory prototype ultrafiltration-adsorption system has two adsorbent channels, each equipped with a hollow-fiber membrane adsorber. Changes in flow path were controlled on this unit by manual three-way valves.
Equilibration: Processing begins by equilibrating the system. The equilibration buffer is formulated to the loading conditions for the chromatography adsorbent. The adsorbent is put off line. Ultrafiltration remains on line during the entire process. Concentration and diafiltration begin coincident with sample introduction. Most small contaminants are eliminated through the permeate during this phase. The chromatography adsorbent goes on line before the retentate is fully equilibrated, and the retentate is recycled through the adsorbent while buffer exchange and concentration continue. The process is complete when buffer conditions in the system match the equilibration buffer.
Monoliths and Membrane Adsorbers: The chromatography adsorbent can be a monolith, a membrane adsorber, or a packed column. Monoliths and membranes have lower protein-binding capacity per unit volume than columns with similar selectivity, but the ultrafiltration step reduces that capacity requirement. Monoliths and membranes, meanwhile, maintain their binding efficiency at flow rates over 10× higher than columns. That has value when integrating ultrafiltration with solid-phase adsorption because the filter area is relatively large and requires high volumetric flow rates to support reasonable process times.
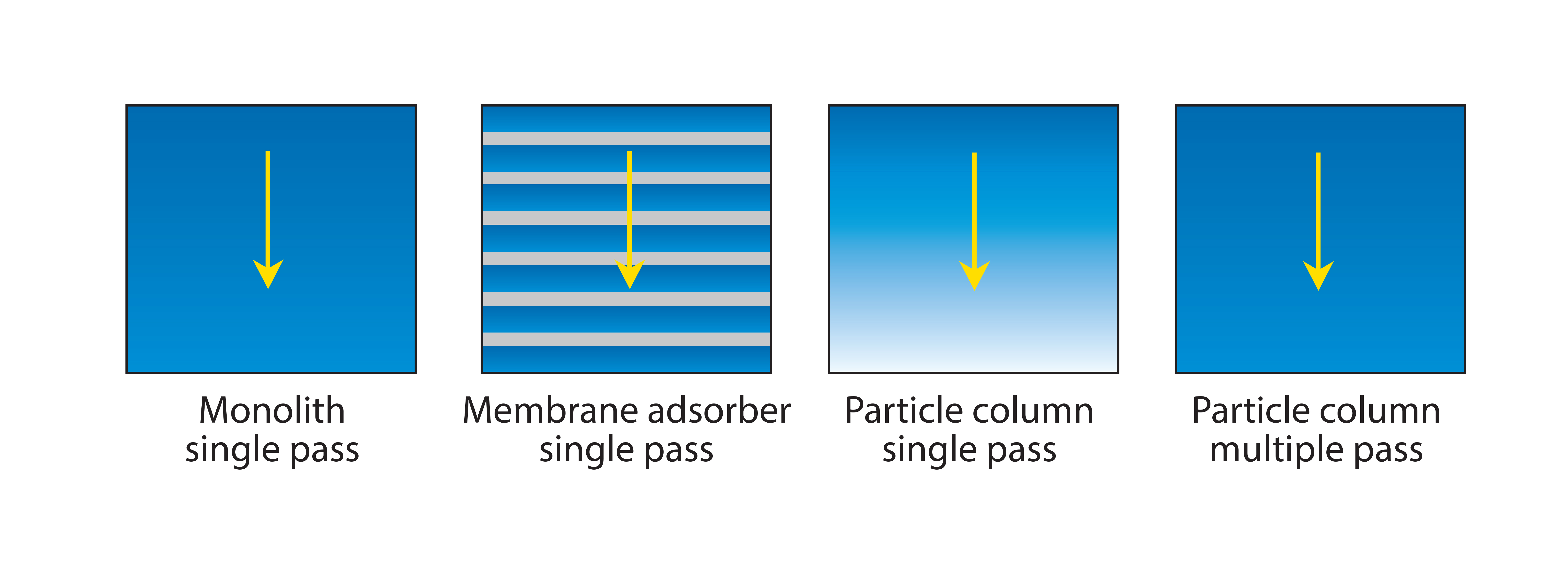
Figure 6: Efficiency of media use by different chromatography formats; arrow shows the direction of flow. Monoliths and membrane adsorbers both achieve virtually complete saturation with only a single pass because mass transport is convective. Columns packed with porous particles seldom achieve much better than 50% saturation in a single pass because of the inefficiency of diffusive mass transport.
Monoliths and membrane adsorbers are also preferred because they support much better media use. Saturation can be achieved with only a single pass because both types of adsorbers rely on convective mass transport. Convective mass transport efficiency is independent of both flow rate and solute size. Columns packed with porous particles rely on diffusion for product to enter the pores, and diffusive efficiency is dependent on both flow rate and solute size. This explains why single-pass loading fails to saturate packed columns in a single pass (Figure 6) and why they benefit from multiple-pass loading.
Porous-particle columns remain an important option despite their slowness because they come with a higher diversity of ligands than monoliths or membrane adsorbers, especially including multimodal adsorbents. Their linear flow-rate restrictions can be compensated for with short beds in either axial or radial-flow formats. Another benefit of a porous-particle column is that its bed volume can be customized to match the capacity requirements of a separation.
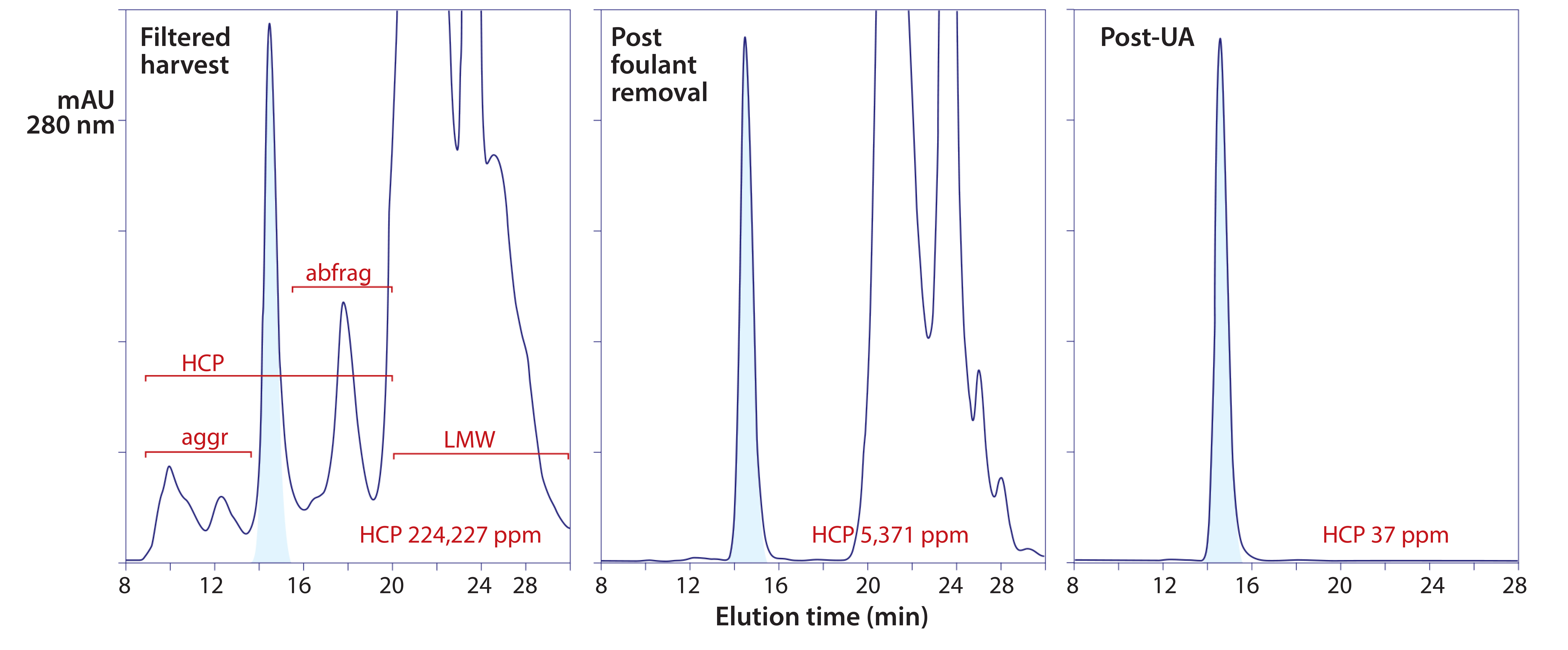
Figure 7: Experimental results from foulant removal, followed by ultrafiltration-adsorption with a single adsorbent channel in flow-through mode; the tangential-flow filtration (TFF) cartridge was a regenerated cellulose membrane with a 30-kDa cutoff. The adsorbent was a strong anion-exchange monolith.
Figure 7 illustrates results from an experiment with a prospective Herceptin (trastuzumab) biosimilar. Chemical foulants were extracted in advance with allantoin, octanoic acid, and electropositive particles or depth filters as described elsewhere (1–5, 14). The buffer was 50 mM Tris, pH 8.0. Antibody was concentrated to 20 g/L over the course of the experiment. The adsorbent was put in line after 2.5 diavolume (DV) of concentration/ buffer exchange, and the experiment was complete at 5 DV. Process time was 4–6 h. HCP were reduced to <37 ppm, DNA to <1 ppb, and aggregates to about 0.1%. Process recovery was 86%.
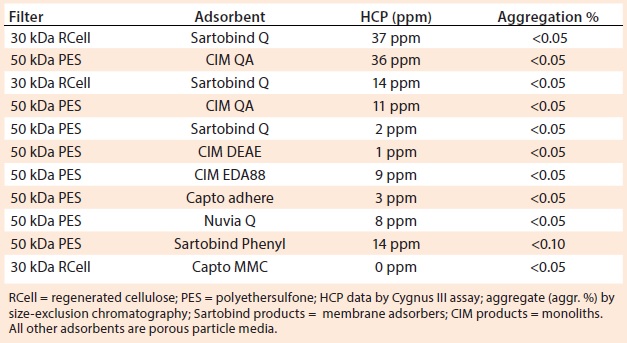
Table 1: Ultrafiltration with a single adsorbent channel in flow-through mode
Table 1 shows results from different harvests of the same cell line using different foulant removal methods and different ultrafiltration media and adsorbents. In brief, 30-kDa cellulose ultrafiltration membranes gave the same results as 50-kDa PES membranes. Membranes with 100-kDa ratings caused antibody losses with this antibody but might be suitable for other antibodies. Monoliths, membrane adsorbers, and porous-particle columns were interchangeable. Strong and weak anion-exchange adsorbents were interchangeable. A multimodal cation exchanger (Capto MMC) and a phenyl membrane adsorber gave results comparable to those of anion-exchangers.
This is not to suggest that the technique will work with any adsorbent for any antibody. Conditions were optimized for each adsorbent. With other antibodies, performance among adsorbents varied more substantially. In all cases, however, performance was affected most dramatically by the efficiency of foulant removal. Omitting foulant removal resulted in universal failure, with HCP contamination commonly persisting at 10,000–30,000 ppm.
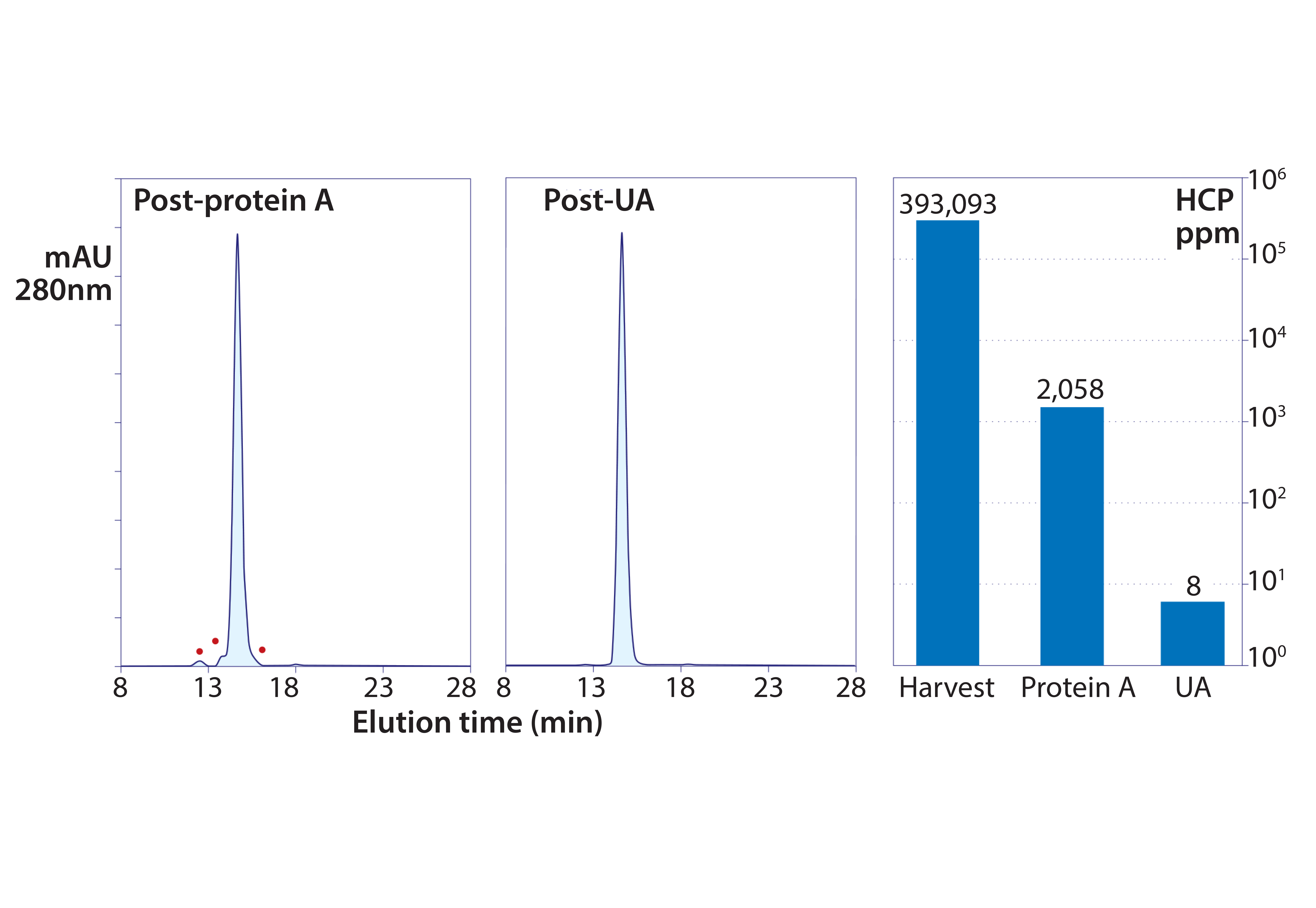
Figure 8: Experimental results from foulant removal followed by capture with protein A, then polishing ultrafiltration-adsorption with a single adsorbent channel in flow-through mode; the TFF cartridge was a PES membrane with a 50-kDa cutoff. The adsorbent was a weak anion exchange monolith (CIM EDA).
Ultrafiltration–Adsorption for Polishing After Stand-Alone Capture
Figure 8 summarizes a purification in which a protein A eluate still at pH 3.5 was introduced to the system. It was concentrated to 25 g/L and buffer-exchanged to 50 mM MES, pH 5.5, while it recycled through a weak anion-exchange monolith. A 0.22-µm membrane filter was placed in line ahead of the monolith to remove turbidity that developed during pH adjustment. This particular antibody was a rare outlier that still contained >2,000 ppm HCP after the protein A step despite having been treated in advance to remove foulants. Coordinated ultrafiltration–adsorption still reduced HCP to <10 ppm, DNA to <1 ppb, and aggregates to <0.1% (13–15).
The ability of the method to reduce HCP from 2,000 ppm to less than 10 ppm highlights two important points: First, by suspending the mechanisms that compromise purification performance, advance foulant removal enables purification methods to achieve the results they should be able to deliver. Second, keeping the ultrafiltration channel in line eliminates small contaminants throughout the entire process cycle. This is an especially important enhancement for flow-through applications because it removes nonadsorbed small contaminants from the system. By contrast, nonadsorbed contaminants in conventional flow-through chromatography applications stay with the antibody.
Coordination of Multistep Chromatography Processes
Flow-through steps with a single adsorbent channel are most compelling: They involve fewer buffers and less process time, but additional adsorbent channels can be added to enable complete multistep purification processes on a single instrument.
For two flow-through channels, the process begins with sample equilibration/concentration. The first adsorbent is put in line and kept there until the sample is equilibrated. Then that adsorbent is put off-line, and the second is brought in-line. The system is equilibrated with a buffer designed to optimize contaminant removal with the second adsorbent. Then rinsing the system with clean buffer displaces product from the internal flow path, after which the system is sanitized with 1 M NaOH.
Using adsorbents in bind–elute mode involves more buffer inputs and takes more time but still conserves the benefits of keeping sample concentrations high throughout the process. It also suspends the difficulty of buffer exchange between process steps. Once your protein is in the system, you can buffer-exchange it in conjunction with adsorption. Large non-IgG proteins such IgM, factor VIII, and von Willebrand factor particularly suggest themselves as natural subjects for ultrafiltration-adsorption because they could accommodate membranes with larger pore-size distributions, potentially removing an even broader spectrum of contaminants.
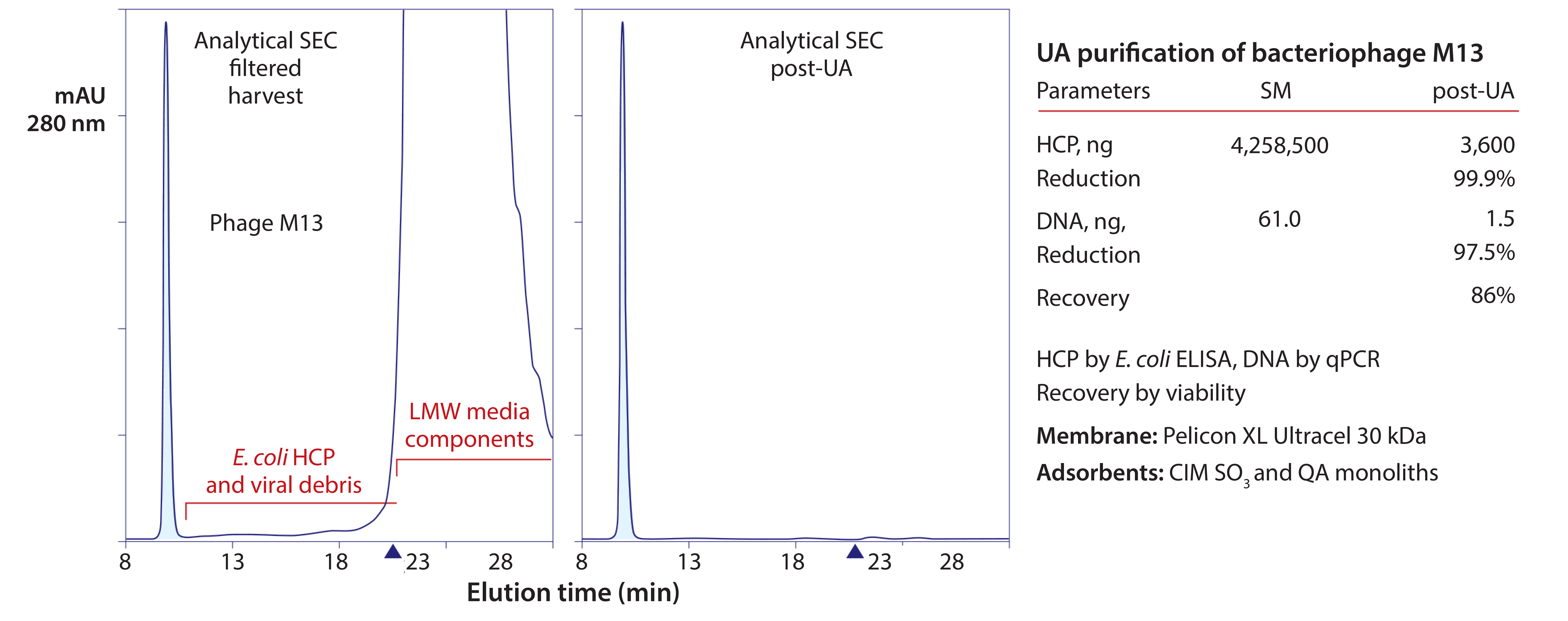
Figure 9: Experimental size-exclusion chromatography (SEC) results showing purification of bacteriophage M13 with a two-channel ultrafiltrationadsorption (UA) system; baseline triangles mark the point at which sample salts elute from the column; ELISA = enzyme-linked immunosorbent assay, qPCR = quantitative polymerase chain reaction, QA = quaternary amine
Virus particles for vaccines, gene therapy vectors, or antibiotic replacement also are obvious candidates. Figure 9 summarizes purification of a bacteriophage with a two-channel apparatus (13–15). The steps were concentration and elimination of small contaminants through the membrane in parallel with equilibration to 50 mM MES, pH 6, then binding to a cation-exchange monolith. The cation exchanger was eluted and put off-line, and an anion-exchange monolith put in-line. The buffer was exchanged to 50 mM HEPES, pH 7.0, causing the virus to bind; then it was eluted.
Future Directions
The biopharmaceutical industry’s evolution toward continuous processing raises the obvious question of whether coordinated ultrafiltration-adsorption can fulfill this ideal. It can, but in a fundamentally different way than approaches such as simulated moving-bed (SMB) chromatography. SMB represents genuine continuous processing: It processes feed continuously at the rate produced and continuously delivers processed product. SMB, however, supports only a single chromatography method per instrument. Multiple instruments are required to conduct multistep processes. Each instrument contains dozens of moving parts and coordinates thousands of mechanical events per day. Method development requires sophisticated simulation software to model the
nonintuitive retrograde order of chromatography events across multiple columns.
Processing with ultrafiltration-adsorption occurs in batch mode, but it can support a complete multistep purification process on a single instrument. All the system’s components can be sanitized ahead of use in a single step and at the end of a process in a single step. A simple surge tank to accumulate harvest between cycles enables round-the-clock production. Even systems configured for multiple adsorption steps involve only a fraction of the moving parts and monitoring devices required by SMB and thousands of fewer mechanical events per process cycle. Development involves established intuitive concepts and familiar guidelines.
Equipment access could be considered a limitation for ultrafiltration-adsorption systems to the extent that they are not presently available commercially. But the components are. Data discussed in this article were obtained with commercially available integrated tangential-flow filtration units, modified to include UV, pH, and conductivity sensors for in-line process monitoring. Those components have been available at a number of process scales for decades, as have the chromatography media, so scalability seems unlikely to be an issue. Experienced chromatographers can assemble a functioning unit within a few hours.
It is impossible to predict whether or when the industry will be ready to embrace another new instrument technology, but several key points are already clear: Ultrafiltration is absolutely capable of providing the processing benefits envisioned by the industry’s early founders, and we can certainly use it more effectively than tradition has taught us. The evolution of chemistry and instrumentation since those days has put us in better-than-ever position to do so. The increasing productivity and economic demands being placed on the industry meanwhile demand that we not overlook opportunities that lie easily within our reach.
Acknowledgments
Thanks to the Bioprocessing Technology Institute in Singapore, where the foulant removal and ultrafiltration-adsorption technology discussed in this article were developed under a grant from the Singaporean Agency for Science, Technology and Research (A*STAR).
References
1 Gagnon P, et al. Nonspecific Interactions of Chromatin with Immunoglobulin G and Protein A, and Their Impact on Purification Performance. J. Chromatogr. A 1340, 2014: 68–78; doi:10.1016/j.chroma.2014.03.010.
2 Gagnon P, et al. Non-Immunospecific Association of Immunoglobulin G with Chromatin During Elution from Protein A Inflates Host Contamination, Aggregate Content, and Antibody Loss. J. Chromatogr. A 1408, 2015: 151–160; https://doi.org/10.1016/j.chroma.2015.07.017.
3 Nian R, et al. Advance Chromatin Extraction Improves Capture Performance of Protein A Affinity Chromatography. J. Chromatogr. A 1431, 2016: 1–7; doi:10.1016/j.chroma.2015.12.044.
4 Nian R, Gagnon P. Advance Chromatin Extraction Enhances Performance and Productivity of Cation Exchange Chromatography-Based Capture of Immunoglobulin G Monoclonal Antibodies. J. Chromatogr. A 1453, 2016: 54–61; doi:10.1016/j.chroma.2016.05.029.
5 Gagnon P, et al. Chromatin-Mediated Depression of Fractionation Performance on Electronegative Multimodal Chromatography Media, Its Prevention, and Ramification for Purification of Immunoglobulin G. J. Chromatogr. A 1374, 2016: 145–155; doi:10.1016/j.chroma.2014.11.052.
6 Tan L, et al. Characterization of DNA in Cell Culture Supernatant By Fluorescence-Detection Size-Exclusion Chromatography. Anal. Bioanal. Chem. 407, 2015: 4173–4181; doi:10.1007/s00216-015-8639-9.
7 Vagenende V, et al. Amide-Mediated Hydrogen Bonding at Organic Crystal Interfaces Enables Selective Endotoxin Binding with Picomolar Affinity. ACS Appl. Mat. Interfaces 5(10) 2013: 4472–4478; doi:10.1021/am401018q.
8 Vagenende V. et al. Self-Assembly of Lipopolysaccharide Layers on Allantoin Crystals. Colloids Surf. B Biointerfaces 120, 2014: 8–14; doi: 0.1016/j.colsurfb.2014.04.008.
9 Vagenende V. et al. Allantoin As a Solid Phase Adsorbent for Removing Endotoxins. J. Chromatogr. A 1310, 2013: 15–20; doi:10.1016/j.chroma.2013.08.043.
10 Singh N, et al. Clarification Technologies for Antibody Manufacturing Processes: Current State and Future Perspectives. Biotechnol. Bioeng. 113(4) 2016: 698–716; doi:10.1002/bit.25810.
11 McNerney T, et al. PDADMAC Flocculation of Chinese Hamster Ovary Cells: Enabling a Centrifuge-Less Harvest Process for Monoclonal Antibodies. mAbs 7(2) 2015: 413–427; doi:10.1080/19420862.2015.1007824.
12 Kang Y, et al. Development of a Novel and Efficient Cell Culture Flocculation Process Using a Stimulus Responsive Polymer to Streamline Antibody Purification. Biotechnol. Bioeng. 110(11) 2013: 2928–2937; doi:10.1002/bit.24969.
13 Gagnon P, et al. Flow-Through. Turbocharged. Oral presentation, Prep XVIII, Philadelphia, July 26–29, 2015.
14 Gagnon P. Apparatus and Methods for Fractionation of Biological Products. United States Patent Application 20170173537, 2017.
15 Gagnon P, et al. The Nokia Syndrome. Can Protein A Survive? Oral presentation, IBC Bioprocess Development and Production Week, Huntington Beach, March 30–April 2, 2015.
Pete Gagnon is CSO at BIA Separations and a member of BPI’s Editorial Advisory Board; pete.gagnon@biaseparations.com.