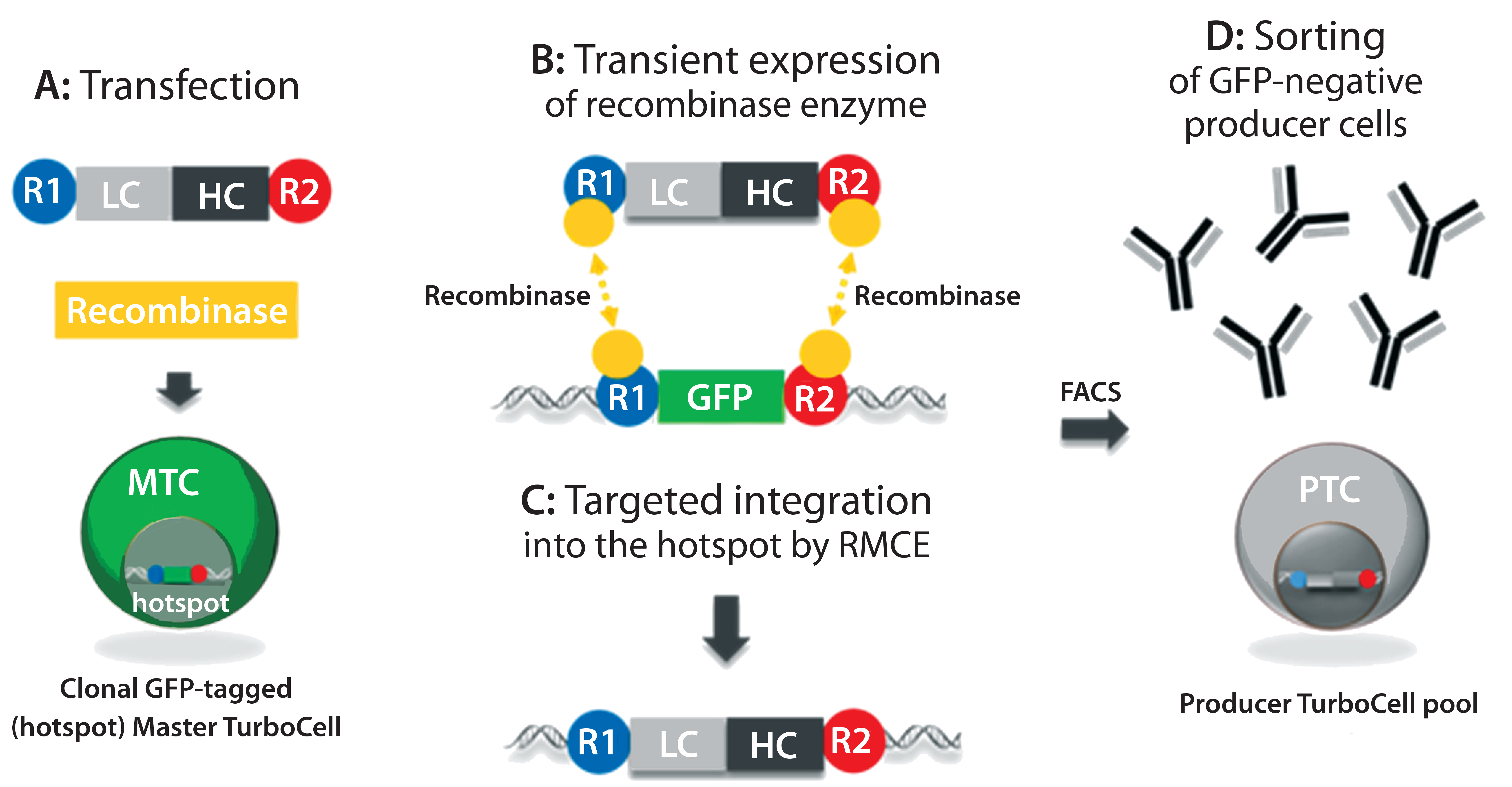
Figure 1: In the TurboCell process of rapid stable cell line development, a target vector containing the gene(s) of interest flanked by recombinase-recognition sites is transiently transfected into the GFP-expressing master TurboCell clone together with an expression plasmid for recombinase (A). After recombinase-mediated cassette exchange (B), GFP expression is lost from the GoI-expressing cells (C), which can be separated from nonrecombinated cell by FACS (D). GFP = green fluorescent protein, FACS = fluorescence-activated cell sorting, GoI = gene of interest.
In the fight against cancer, antibody–drug conjugates (ADCs) represent an increasingly important therapeutic approach. These biopharmaceuticals are designed to maximize the therapeutic index of cytotoxic small-molecule drugs through their selective delivery to tumor cells while leaving normal, healthy cells untouched. Structurally, an ADC is a monoclonal antibody (MAb) conjugated by a chemical linker to a potent cytotoxic drug. Conceptually, the MAb serves as the delivery component, targeting a specific tumor antigen that ideally is not expressed (or is expressed at very low levels) on normal cells. Following internalization of the conjugated complex, the free toxin is released to kill the tumor cell (1).
Table 1: Candidate antibody–drug conjugates (ADCs) and their characteristics
Product development represents a major obstacle to delivering these new therapies to patients. The industry is striving constantly to streamline operations, reduce timelines, and minimize risk to increase product development success. We have developed a “gene to ADC product” strategy to fast-track ADC production and lead selection. Our approach has been validated using ADCs based on trastuzumab (TMAb), targeting the human epidermal growth-factor receptor 2 (HER2). We selected four TMAb-based ADCs with different antibody formats and conjugation approaches (in the toxin-linker design and properties of the circulating ADC and released toxin) as representative of a potential set of lead candidates in an ADC product-development project (Table 1).
In selecting specific ADCs, we compared two factors: locations of the conjugated linker-toxin and cleavability of the linkers. With the former, stochastic conjugation to TMAb with up to eight potential thiol sites was compared with site-specific conjugation using Thio-TMAb (TMAb LC V205C), which has two free thiol sites on its light chain. With the latter, the selected linkers give rise to metabolites that have different properties. The cleavable linker liberates a cell-permeable toxin that can exhibit a bystander effect on antigen-negative cells. However, it is also a substrate for multidrug resistance (MDR) exporters and has limited efficacy in cells with high pump expression and activity. By contrast, the noncleavable linker liberates a toxin that has no cell permeability or bystander effect, but is active in MDR-positive cell lines.
MAb Production Process
We produced TMAb and Thio-TMAb LC V205C to evaluate stochastic and site-directed conjugation, respectively. Each MAb was expressed by a Rentschler Turbo Cell modified CHO K1 host cell line using expression vectors that contained independent expression units for the light chain (LC) and heavy chain (HC), both controlled by a cytomegalovirus (CMV) promoter and terminating SV40 poly-A signal (Figure 1) (2). For Thio-TMAb, we introduced a single cysteine mutation (V205C) into the TMAb LC gene using the Q5 site-directed mutagenesis kit and specific primers. TMAb and Thio-TMAb expression vectors each were transfected into host cells together with an expression plasmid for the site-specific recombinase. After recombinase-mediated cassette exchange had occurred — resulting in the loss of green fluorescent protein (GFP) reporter expression — we isolated transfected cells using fluorescence-activated cell sorting (FACS).
After 3.5 weeks of expansion, antibody-producing cell pools were cultivated in a generic fed-batch process. Once seeded (0.3 × 106 cells/mL) in chemically defined medium, they were successively fed on days 3, 5, and 7. We cultured the cells in shake flasks at 37 °C until day 7, then shifted them to a reduced temperature of 33 °C until harvest at day 17. That cell culture harvest was clarified by depth filtration (DF) followed by normal-flow filtration through a 0.22-µm membrane.
We purified our MAbs by protein A chromatography followed by viral inactivation, which involved holding the eluate at pH 3.6 for 75 minutes with subsequent neutralization to pH 6.0. Then we formulated them by ultrafiltration/diafiltration (UF/DF) with a 30-kDa nominal molecular-weight cutoff (NMWCO) at 15 g/L in 5 mmol/L of histidine–HCl and 50 mmol/L trehalose at pH 5.9. A post-DF solution was spiked to a final concentration of 0.01 % (w/v) polysorbate-20. Within five weeks, we generated cell pools that consistently expressed TMAb or Thio-TMAb and expanded them to provide 8 L of harvest supernatant, yielding ~2 g formulated material sufficient for the early phase studies.
Table 2: Summarized protein analysis results for MAb and Thio-MAb before conjugation
To ensure that those MAbs were suitable intermediates for subsequent conjugation with linker-toxin, we evaluated their molecular integrity and purity. For nonreducing sodium-dodecyl sulfate (SDS) capillary electrophoresis (CE), we used a LabChip Protein Express 200 assay from Perkin Elmer. Samples were denatured at 70 °C for two minutes using the kit-provided sample buffer spiked with 10 mmol/L N-ethylmaleimide. Results demonstrated that both MAbs were intact molecules with minimal levels of smaller fragments and free HC or LC (Figure 2, Table 2).
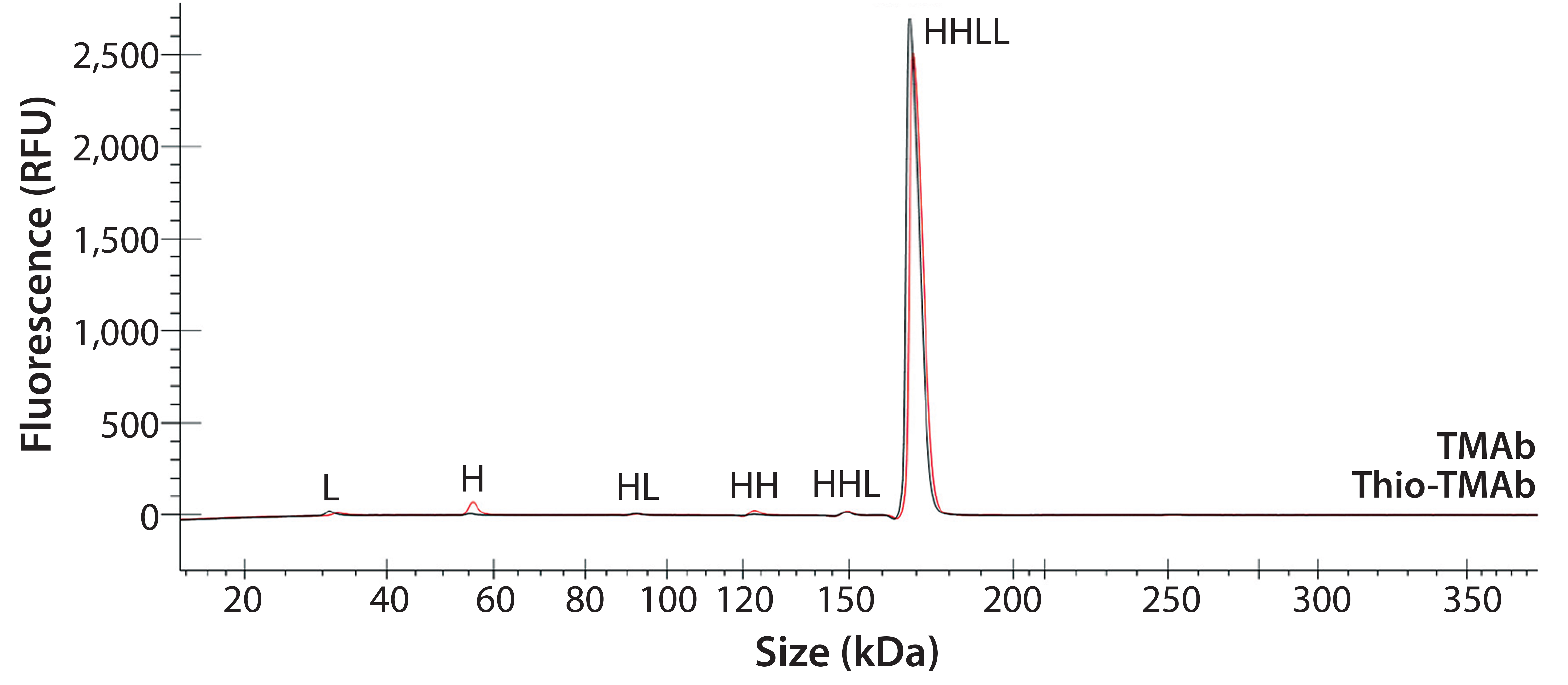
Figure 2: Electropherogram overlay obtained from nonreducing LabChip protein assay (Perkin Elmer); black = TMAb, red = Thio-TMAb; H = heavy chain, L = light chain
We detected higher levels of free HC in the Thio-TMAb formulation; however, because the intact levels were >96%, we deemed those to be insignificant for our early phase development activities. A potential concern with such cysteine-engineered MAbs is that they can form triple-LC variants by forming nonnative disulfide bonds between free LC thiols and full-length MAbs (3). No such variant impurities were detected on the Thio-TMAb nonreducing SDS electropherogram, however (Figure 2).
TMAb and Thio-TMAb differ only in the latter’s LC V205C site mutation, which theoretically is charge neutral (4). Certainly, isoelectric focusing (IEF) revealed pI values for both MAbs similar to the theoretical pI = 8.4, with both antibodies showing three basic bands and three or four acidic bands. A LabChip charge-variant assay (Perkin Elmer) revealed similar charge distribution profiles for both MAbs, although the Thio-TMAb showed a significantly higher content of acidic isoforms (47%) than the nonengineered TMAb (29%) (Table 2). We believe that phenomenon to be caused by cysteinylation and/or glutathionylation at the engineered cysteine on the LC, altering the surface-charge distribution of the Thio-MAb (5). We did not expect such chemical modifications to free thiol groups on Thio-TMAb to affect the conjugation chemistry.
For aggregation analysis by size-exclusion high-performance liquid chromatography (SE-HPLC), we ran a TSKgel G3000SWXL column (Tosoh Bioscience) at a flow rate of 1 mL/minute using 100 mmol/L phosphate and 100 mmol/L sodium chloride at pH 7.2. Results for both MAbs revealed three high–molecular-weight (HMW) components. The monomer level of the nonengineered TMAb was 96.5%, which is common for antibody production at cell-pool level (6). For the Thio-TMAb monomer content, a value of 92.4% was obtained. For the most part, that appeared to be related to an increased content of HMW3 material, which is the smallest of the HMW components. With the Thio-MAb, HMW3 comprised 4% of the pool compared with 0.8 % for the TMAb. Our nonreducing SDS electropherograms showed no HMW species for either MAb (Figure 2), so we consider the detected HMW SEC peaks to be noncovalent aggregates.
MaB conjugation Process Following production of the MAb intermediates and analysis to demonstrate that those materials are suitable for further development, the second stage of our fast-track process is conjugation chemistry. We intended to produce TMAb ADC variants for evaluation in a lead-selection exercise. Several approaches have been developed to conjugate toxins to MAbs (7). For this study, we selected the classical cysteine conjugation approach using maleimide chemistry. When combined with partial interchain disulphide bond reduction, it yields eight potential stochastically distributed sites for linkage and linker-toxins. With Thio-TMAb, the incorporation of the free thiol groups provides for site-specific conjugation to LC 205C.
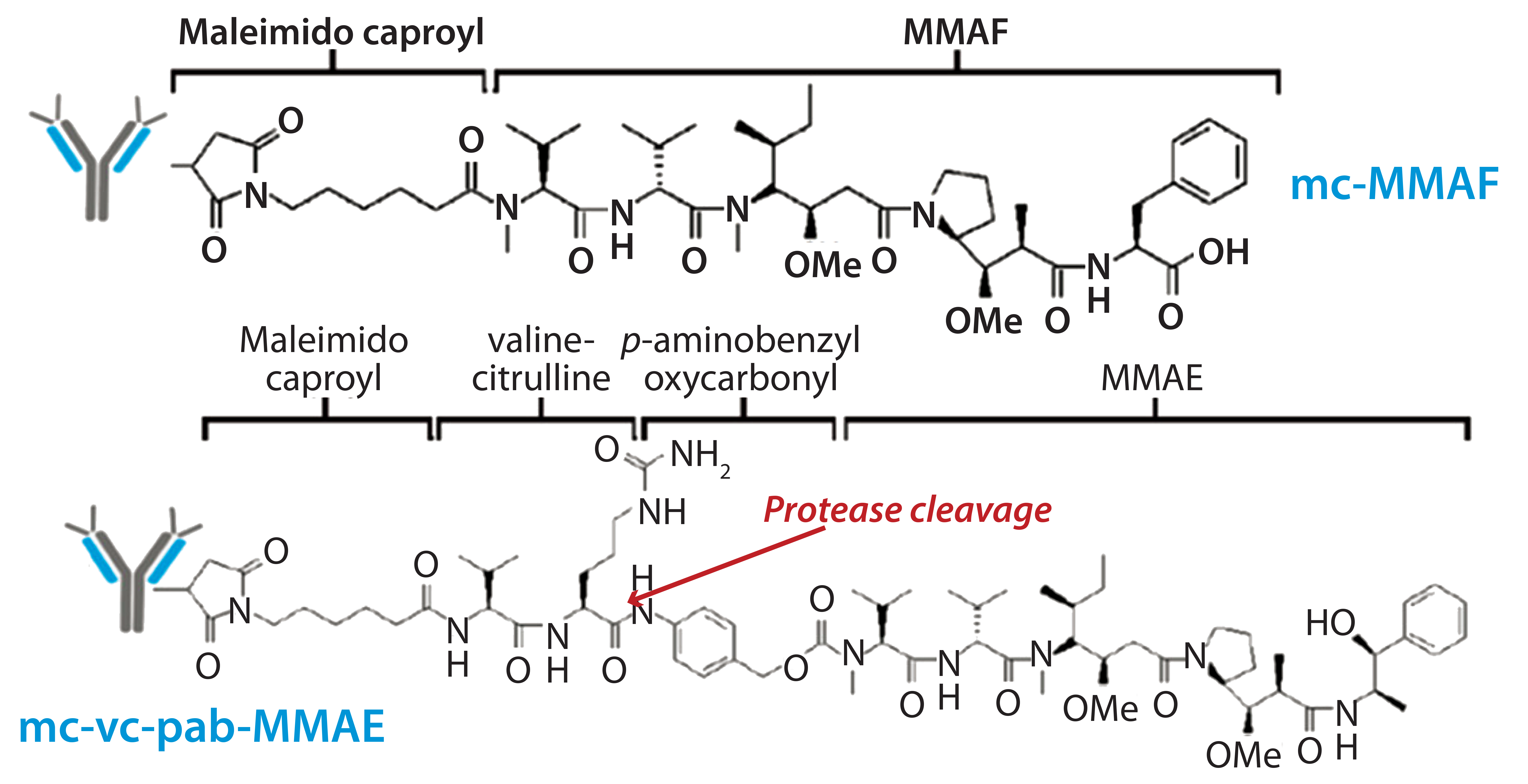
Figure 3: Schematic diagram shows the chemical structures of ADC linker and toxins.
The other structural component this study was intended to evaluate is that of linker-toxin. We chose two tubulin-inhibiting auristatin derivatives (Figure 3):
- the cathepsin B cleavable maleimidocaproyl valine-citrulline-p-aminobenzyl-auristatin E (vcMMAE)
- uncleavable maleimidocaproyl-monomethyl auristatin F (mcMMAF).
Regardless of the type of conjugate and conjugation approach involved, ADCs share two common critical quality attributes (CQAs) specific to this format of biologic: the average ratio of drug to antibody (DAR, including the pattern of drug loading and the level of unconjugated antibody) and the level of residual process-related impurities including both the toxin-linker and conjugation cosolvent. One method for determination of DAR is hydrophobic-interaction chromatography (HIC) HPLC, which fractionates intact ADCs based on increased hydrophobicity with increasing toxin conjugation. By determining the peak area for each ADC peak, it is possible to derive an average DAR value.
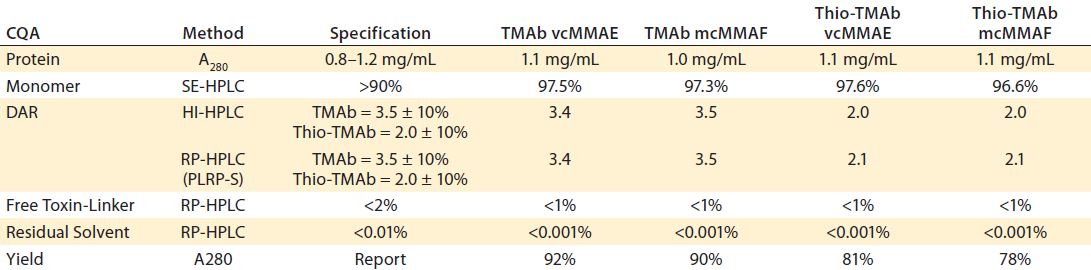
Table 3: Target specification and quality achieved following minimal development studies; DAR = drug/antibody ratio, SE = size exclusion, HIC = hydrophobic interaction, HPLC = high-performance liquid chromatography, RP = reversed phase; PLRP-S is a proprietary RP resin of Agilent Technologies.
Another technique using the same principle to evaluate the LC and HC components after reduction is reverse-phase (RP) HPLC. Together, the two orthogonal techniques provide complementary information on average DAR as well as the pattern and position of conjugation. Similarly, process-related impurities and residual solvent are determined by specific RP-HPLC assays. The ADC’s intended use drives the specification for each of these specific quality attributes. For the purposes of our screening and lead-candidate selection project, we established a target specification and applied platform analytical techniques (Table 3).
Our initial approach was to achieve target specification without extensive process development effort. The preliminary conjugation experiments are designed around platform processes initially for the common stochastic and site-specific conjugation methods. Typically they are performed on a scale of 1–5 mg. In addition to the specification assays, we used nonreducing SDS polyacrylamide gel electrophoresis (SDS-PAGE) to provide qualitative information on the pattern of reduction for stochastic TMAb conjugations and confirm the fidelity of reoxidation for Thio-TMAb conjugations.
Regarding the conjugation chemistry itself, generally it follows a two-phase empirical approach. The first phase is a general screen of the platform reduction/conjugation conditions; the second phase is an optimization process. Although the basic maleimide chemistry was used both for the stochastic and site-specific conjugations, we required different approaches for the two ADC variants.
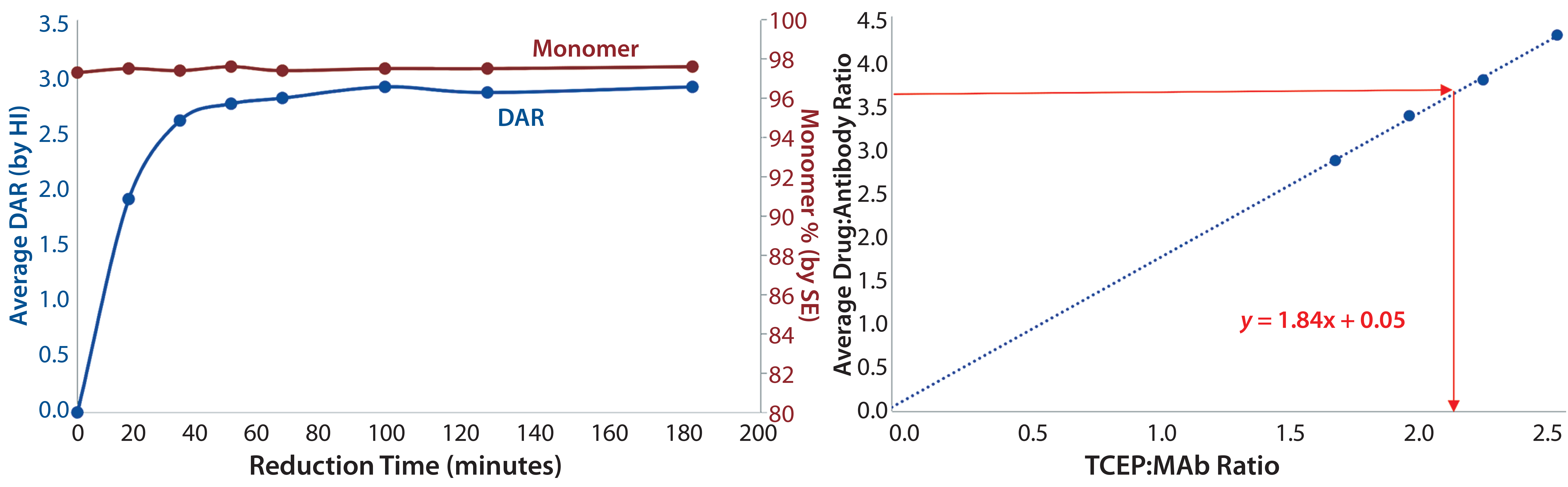
Figure 4: (left) Relationship between reduction time and average DAR and monomer at 1.5 moles of TCEP per mole of MAb; (right) relationship between TCEP:MAb ratio and DAR following 90-minute reduction and alkylation with toxin and linker; the TCEP level required for a target average DAR can be calculated from the linear fit (equation shown in red) of the data or by simply reading from the graph (lines shown in red).
TMAb ADC Development: To prepare TMAb stock for reduction, we adjusted the pH to 7.5 and added ethylenediaminetetraacetic acid (EDTA) to prevent metal-catalyzed reoxidation. Reduction was initiated by adding Tris(2-carboxyethyl) phosphine (TCEP) at 1.5 moles per mole of antibody, then the mixture was stirred at 20 °C for three hours. We chose TCEP as the reductant for this process because it contains no thiol and does not interfere with the subsequent protein thiol and linker-toxin maleimide conjugation. We monitored the extent of reduction over time by sampling the reaction and alkylating the free thiols generated with vcMMAE or mcMMAF added at 5 moles per mole of antibody from a 10 mmol/L solution in dimethylacetamide (DMA) with additional DMA to achieve a total 10% (v/v) DMA cosolvent. Analysis of the resulting conjugates by HIC-HPLC and SEC-HPLC revealed that the platform reduction reaction was complete after 90 minutes, as exemplified by production of TMAb-vcMMAE (Figure 4, left panel).
Although the ADC products had not achieved their target DARs of 3.5, our desired quality attributes were met. Aggregation did not increase. We optimized the process by evaluating the TCEP:MAb ratio. A series of reduction trials at TCEP:MAb ratios between 1.5 and 2.5 revealed that a ratio of 1.9 was required to achieve the target DAR (Figure 4, right panel). Because the chemistry was essentially the same, the optimized conjugation conditions applied to both TMAb-vcMMAE and TMAb-mcMMAF production.
Thio-TMAb ADC Development: As described above, manufacture of the Thio-TMAb appeared to result in cysteinylation and/or gluthionylation of the free thiol groups, which is a common feature during cell expression. Those thiol “caps” must be removed before an engineered cysteine can be conjugated. That is typically achieved through full reduction of an antibody using mild conditions that reduce the interchain and engineered thiols but not the domain-stabilizing intrachain disulphides. All free reductant/capping thiols are removed before the interchain disulphides can reform by a reoxidation process. Dithiothreitol (DTT) is a good reductant typically used for this process because, despite being a thiol-containing molecule, its removal along
with the capping cysteine/glutathione presents no difficulty in the subsequent reoxidation and conjugation steps.
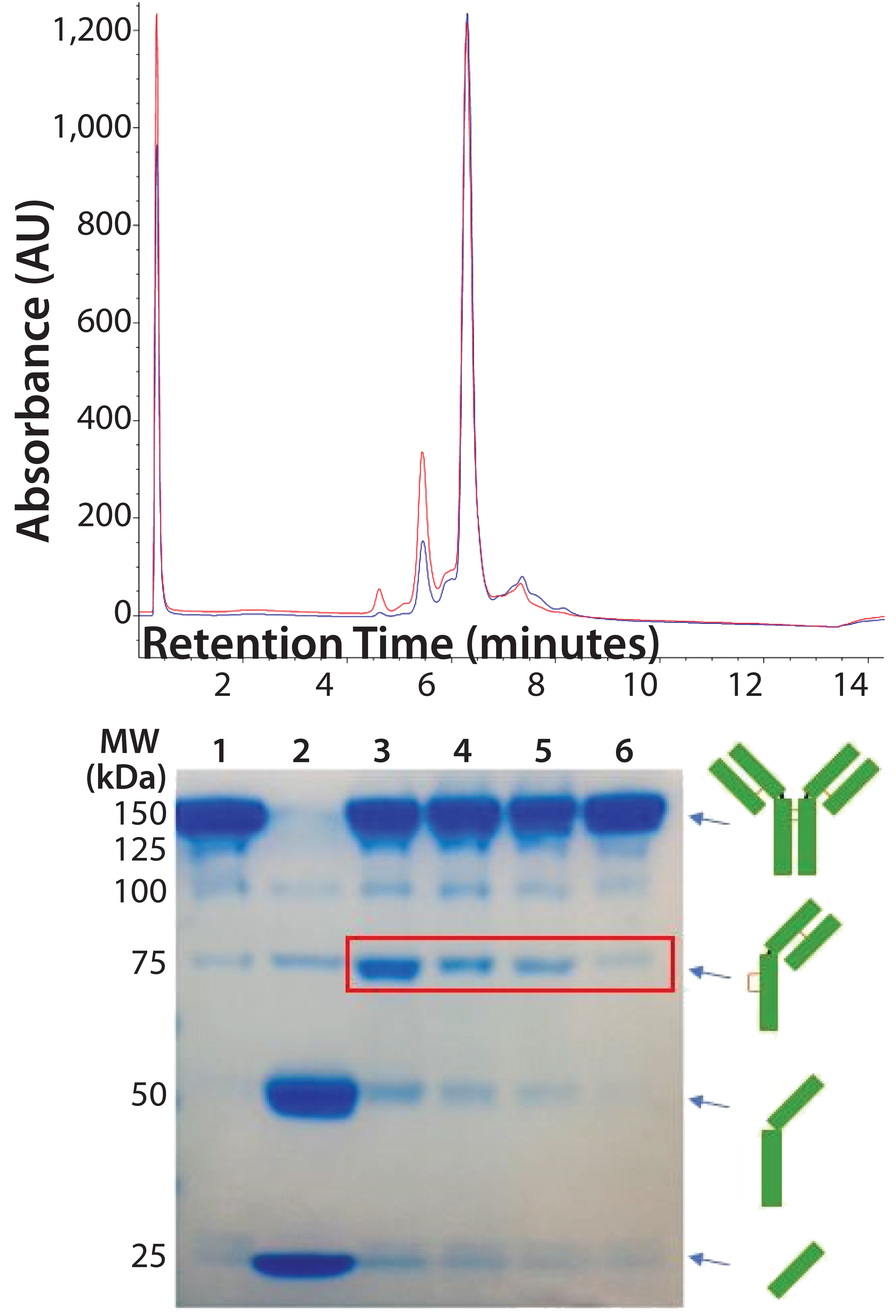
Figure 5: (top) HIC analysis shows improvement in the extent of conjugation at pH 7.0 (blue line) relative to pH 7.5 (red line). A single sample of reduced and reoxidized Thio-TMAb was split in two and conjugated under identical conditions except for the conjugation pH. (bottom) 8–16% nonreducing SDS-PAGE of Thio-TMAb (Lane 1), reduced Thio-TMAb (Lane 2) and reoxidized Thio-TMAb at pH 8.0 (Lane 3), at pH 7.0 (Lane 4), at pH 6.5 (Lane 5) and at pH 7.0 with five sequential additions of dehydroascorbic acid (Lane 6). The red box highlights the undesired reoxidation pattern.
We initiated our “decapping” reduction by adding DTT at 20 mol per mole of antibody following adjustment of the solution to pH 7.5 and addition of EDTA. The resulting mixture was incubated at 20 °C for 16 hours, having been initially mixed thoroughly and then left unstirred to prevent shear-induced aggregation. Afterward, DTT was removed and the buffer was exchanged for 0.05 mol/L Tris-HCl at pH 7.5, then complete reduction was confirmed by nonreducing SDS-PAGE (Figure 5, bottom panel, Lane 2). We reoxidized the antibody by adding dehydroascorbic acid (dHAA) at 10 mol per mole of antibody and incubating the solution at room temperature for two hours. Finally, we alkylated the free engineered thiols with vcMMAE or mcMMAF (added at 4 mol per mole of antibody in DMA).
Under those initial platform conditions, the extent of conjugation was suboptimal (below the target
DAR of 2). Given that we used forcing conditions with excess toxin-linker, cosolvent, and the Thio-TMAb already in a suitably reduced state, we considered the pH to be the most likely parameter for optimizing the extent of conjugation. Screening different pH levels in the 6.5–8.0 range identified pH 7.0 as optimal for conjugation, which improved the extent of conjugation to yield a target DAR of 2 (Figure 5, top panel).
We also noted that lowering the pH reduced the amount of intrachain disulphide-bond formation, reducing the level of “half-antibodies” measured by nonreducing SDS-PAGE. By controlling the rate of reoxidation at the optimal conjugation pH (adding dHAA in five discrete aliquots, 30 minutes apart, followed by a further 30 minutes of reoxidation), we further reduced half-antibody formation (Figure 5, bottom panel). These conditions applied to both vcMMAE and mcMMAF variants.
R&D Scale ADC Manufacture: Using the two optimized conjugation processes, we produced both stochastic and site-specific ADCs, each with cleavable and uncleavable linker variants, at 100-mg scale. Residual toxin-linkers, solvents, and other process-related impurities were removed, then ADCs were formulated by buffer exchange into 5 mmol/L histidine-HCl, 50 mmol/L trehalose, and 0.01% (w/v) polysorbate-20 at pH 6 using a Vivaspin 30-kDa MWCO polyethersulfone (PES) membrane spin filtration device from Sartorius Stedim Biotech.
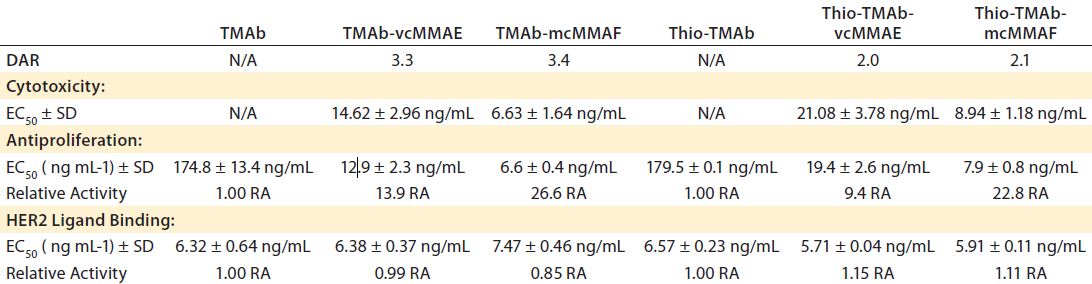
Table 4: HER2 ligand binding, cytotoxicity, and antiproliferative bioactivity of TMAb and Thio-TMAb ADCs are compared with those of their parent MAbs alone.
We tested all four ADCs against the prescribed release specification (Table 3), finding them to meet all CQAs (Table 4). Note that the DAR values were within prescribed specifications, with the Thio-TMAb ADCs being fully conjugated and the stochastic TMAb ADCs having an intended partial conjugation (~3.5 of eight potential sites). The average yield for these four ADCs was 85% (range 81–92%), with a slightly lower yield for the Thio-TMAb conjugates, which was the result of process losses during the additional purification required between reduction and reoxidation/conjugation.
Biological Activity and Developability Risk Assessment
Conjugation chemistry represents the final phase of ADC manufacturing to produce R&D material for lead-candidate selection. Using a fast-track approach for selection of specific MAbs and provision of their respective genes, we produced purified developmental MAb intermediate within four weeks. Development, optimization, and manufacture of the ADCs took a further four weeks. The next phase of our fast-track approach involved determining the biological activity of the ADCs to confirm potency, then a developability risk assessment to evaluate their robustness for further development and manufacturing.
HER2 Binding and Biological Activity: Ligand binding is a CQA for ADCs. We determined this using an indirect enzyme-linked immunosorbent assay (ELISA) with recombinant human HER2. The results revealed that these ADCs were not notably different in HER2 binding from their naked MAbs (Table 4). Consequently, linker-toxin
conjugation to the antibodies by their cysteine groups did not appear to affect the CDR regions significantly.
TMAb has antiproliferative activity, but it is not cytotoxic in itself. As a result, we decided to test for antiproliferation and cytotoxicity, comparing the biological activity of the ADCs with that of their parent MAbs. We determined those activities using a multiplexed cytotoxicity and antiproliferation assay, using the high-HER2–expressing SK-BR3 (HER23+) breast cancer cells as the target. Cytotoxicity was determined by lactate dehydrogenase (LDH) release and proliferation using Alamar Blue vital dye.
All four ADCs were found to be cytotoxic to SK-BR3 cells with high potency, whether the conjugation was stochastic or site-directed. Moreover, the uncleavable mcMMAF variants were significantly more potent than the cleavable vcMMAE forms (Table 4). Because the parent TMAb and Thio-TMAb antibodies were not cytotoxic, these data demonstrate that the bioactivity was attributable to the toxin component. Inhibition of proliferation also was seen with all four ADCs and with the parent TMAbs and Thio-TMAbs; however, the ADCs were significantly more potent than the MAbs, demonstrating greater than ninefold increase in such activity. The auristatin toxins are potent antitubulin inhibitors, so this result is not surprising, although it does demonstrate that these ADCs both inhibit proliferation and induce cell death in their target cells.
Developability Risk Assessment: As outlined in ICH guidance, quality by design (QbD) is the current paradigm for drug development (8). It emphasizes prior knowledge and sound science as a basis for product development and relies on extensive use of risk-management tools to minimize risk and increase the chance of development success (9). Developability risk assessment is one such experimental tool that takes into account a broad range of parameters in assessing a biopharmaceutical for development. Essentially, it is a stress test for evaluating the main routes of protein denaturation and determining protein robustness as a prelude to process and formulation development. When used in a lead-selection process, this can be used to provide prior knowledge and rank the risks related to which molecule is most suitable for the rigors of process and formulation development (and which were the most likely to be problematic).
In the final phase of the “gene to ADC product” approach, we performed a developability risk assessment of the four ADCs to rank their risk for subsequent process and formulation development. The “Risk Assessment” box lists stress conditions used for this study.
| Risk-Assessment Stress Conditions |
| Acid (pH 3, 48 hours at 40 °C)
Base (pH 9, 48 hours at 40 °C) Deconjugation (0.1 mmol/L cysteine, 24 hours at 37 °C) Freeze–thaw (×5) Light (ICH guideline) Oxidation (0.05% tert-butyl hydroperoxide, two hours) Shear stress (pipetting ×50) Thermal degradation (four weeks at 40 °C) |
Those conditions generally are standard for assessment of antibody stability; however, for this particular developability study, we included deconjugation/redox induced loss of toxin. That is specific for cysteine-conjugated ADCs and is designed to test the stability of the conjugation, breakdown of which leads to linker and/or toxin release. Following stress testing, we analyzed the ADCs and unstressed controls using the following methodologies to assess the critical quality attributes: HIC-HPLC for ADC hydropathy and DAR; PLRPS-HPLC (a form of RPC proprietary to Agilent Technologies) for light and heavy chain hydropathy and DAR; cIEF for charged variants; SE-HPLC for multimers, aggregation, and hydrolysis; and intrinsic fluorescence for higher-order structure.
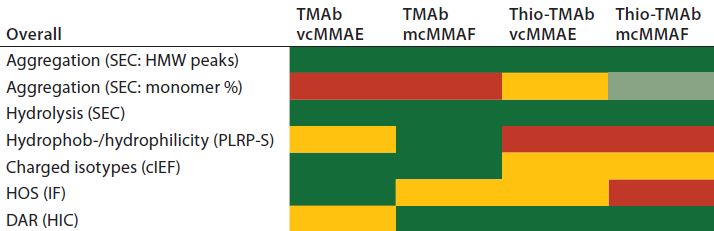
Table 5: Traffic-light schematic shows the developability risk assessment for the overall stability of the TMAb and Thio-TMAb ADCs, highlighting potential routes of degradation.
Although our selection of analytics is not fully comprehensive, we chose those methods to evaluate the conjugates’ CQAs, and together they represent a typical developability risk assessment. After assessing the effects of those stress conditions on the different CQAs, we summarized the results in a series of “traffic-light” diagrams, listing the ADC responses as high, medium, or low (Table 5). In making this assessment, note that in general the stress conditions are designed to be more extreme than would be experienced during a manufacturing process. Nevertheless, they do indicate where there is susceptibility to stress conditions and consequently help us identify the routes of degradation. For brevity, only the salient points of the developability study are discussed below.
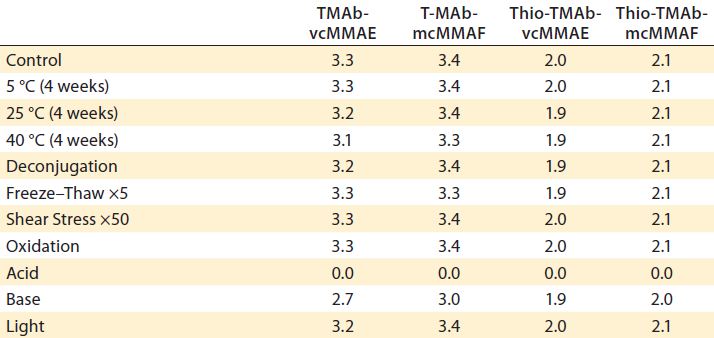
Table 6: DAR determination by hydrophobic-interaction high-performance liquid chromatography (HI-HPLC)
Stability of the linker-toxin is a critical control attribute for all ADCs. We used HIC-HPLC to evaluate this parameter by measuring the DAR (Table 6). Apart from base treatment of TMAb vcMMAE, we found no gross evidence of deconjugation even with cysteine treatment, which was designed specifically to evaluate the cysteine-conjugation stability. Overall this demonstrated linker-toxin stability with three of the four ADCs. Acid treatment did decimate the DAR value, but HIC chromatograms revealed that it heavily degraded the proteins themselves, so it was not due to any specific linker-toxin stability.
We attempted DAR analysis of the ADC LC and HC components using the RP-HPLC method with a polymeric PLRP-S column. It was evident from the resulting chromatograms that this method also was stability-indicating with regard to chemical modifications that would cause hydrophobic and hydrophilic changes to the LC and HC. Such hydropathic modifications were evident in all of the ADCs to varying extent. Consequently, it was impossible to assign DAR values unequivocally to the peaks based on retention time — so we did not attempt DAR analysis by this method. On the other hand, the method demonstrated that the Thio-TMAbs especially were sensitive to stress conditions. The Thio-TMAb mcMMAF ADC in particular underwent hydropathic modifications to both LC and HC with all the stress conditions.
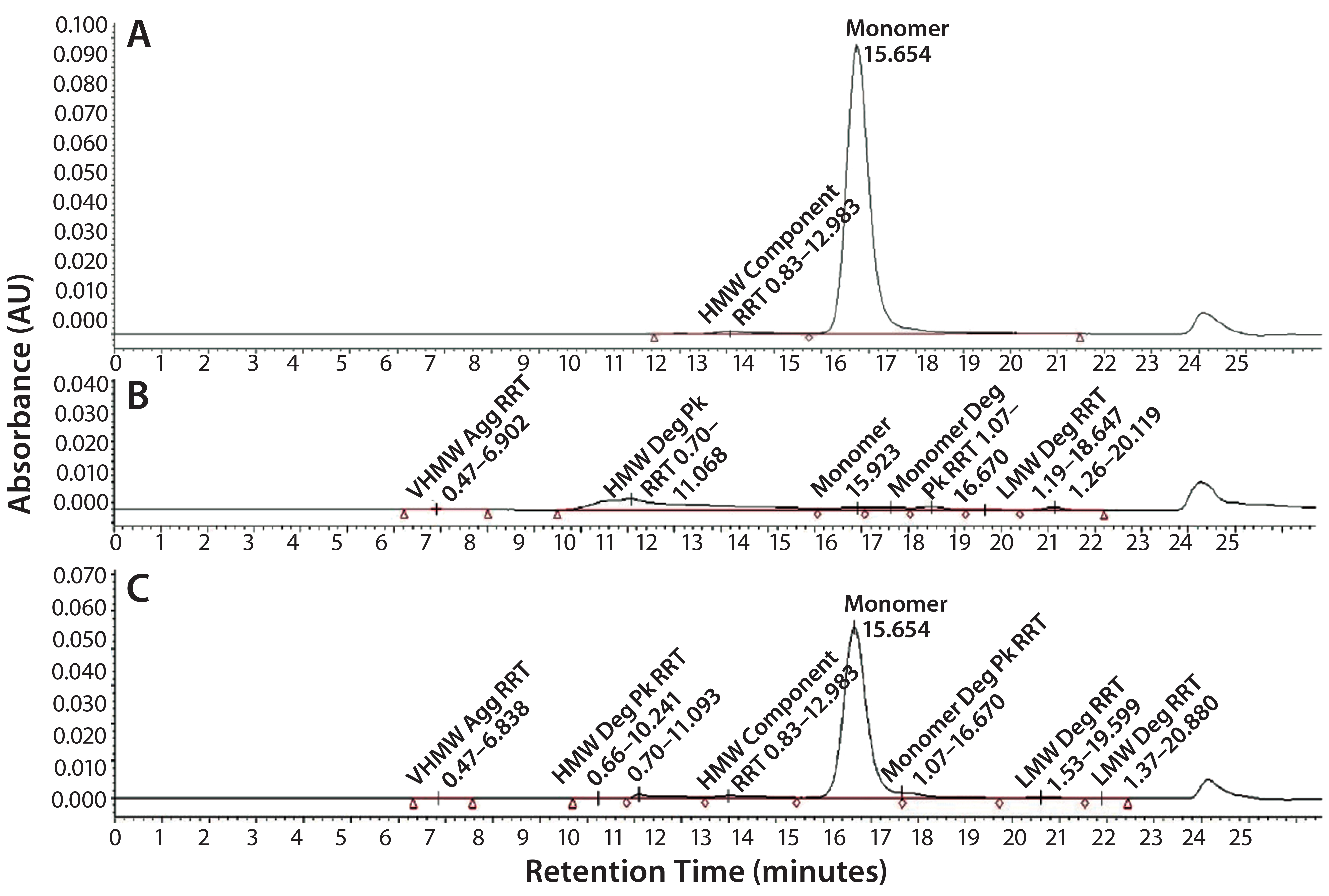
Figure 6: SE-HPLC chromatograms of TMAb vcMMAE; control (A), acid treatment showing total loss of monomer (B), and base treatment showing partial loss of monomer (C).
It is well recognized that the act of conjugating highly hydrophobic linker-toxins to antibodies can destabilize their protein structure, making the molecules more prone to aggregation. Accordingly, we used SE-HPLC as a basic method to evaluate the level of stress-induced aggregation. For simplicity, we used it in two ways. We used the standard approach to evaluate soluble aggregates and dimers by evaluating the HMW materials evident in the chromatogram. And we evaluated insoluble aggregates by determining loss of monomer content resulting from insoluble aggregate particles that are too large to load on the SE column. SE-HPLC analysis of the stressed samples revealed only minimal levels of soluble HMW in all four ADCs, the majority of which corresponded to dimer. In contrast, all the ADCs appeared to be highly prone to stress-induced insoluble aggregate formation, evident by the aforementioned loss of monomer with no parallel increase in soluble HMW forms (Figure 6). The ADC that was least prone to aggregation was the Thio-TMAb vcMMAE variant.
SE-HPLC also can be used to evaluate protein hydrolysis. In our study we scanned the SE chromatograms also for evidence of low–molecular-weight (LMW) material indicating protein breakdown. Apart from acid and base treatments, which caused significant breakdown of all the ADCs, only minor levels of LMW were observed with the stress conditions. Hence, protein hydrolysis did not appear to be a major degradative pathway for these molecules.
To evaluate charged variants and deamidation isotypes, we used cIEF analysis. Following stress testing, no appreciable change in charged variants was observed apart from the base conditions, which would be expected to induce deamidation. Here both the acid and thermal stress conditions induced changes in the cIEF profiles, but they were indicative of marked protein breakdown rather than charge-related modifications such as deamidation (data not shown).
Although it was evident from the SE data above that the conjugation of hydrophobic linker-toxins to the antibodies made the molecules more prone to aggregation, this also raised the question as to whether the conjugations were affecting the basic conformational stability of the ADCs. To evaluate this, we have used intrinsic tryptophan fluorescence as a simple, high-throughput technique for assessing higher order structure (HOS). Tryptophan fluorescence is very sensitive to solvent polarity and local protein environment, hence unfolding of the protein results in a red-shift of the emission spectrum, which was determined using the F350/370-nm ratio as the endpoint. Note that the HOS data indicated that the Thio-TMAb ADCs were less stable than their TMAb counterparts. Additionally, the mcMMAF noncleavable variants were less stable than the vcMMAE ADCs with regard to HOS. This may be due to the increased hydrophobicity of the mcMMAF linker-toxin combination destabilizing the molecule. Indeed, the Thio-TMAb- mcMMAF was shown to be the least structurally stable molecule of the four ADC variants.
Risk Assessment Conclusions: Developability risk assessment is meant to evaluate the robustness of a biopharmaceutical over a range of parameters with appropriate stability-indicating analytical methods. Although our selection of analytics is not fully comprehensive, the chosen methods are platform assays that can provide basic assessment of protein stability in a short time. Thus the developability risk assessment could be performed within eight weeks, with the four-week thermal stress testing representing the major time constraint.
Nevertheless, based on our assessments, it is possible to rank the four TMAb-derived ADCs for robustness with regard to subsequent process and formulation development. Table 5 distills the datasets to provide an overall risk assessment of the propensity of each molecule to undergo denaturation. From that traffic light risk assessment, an overall risk assessment for lead selection becomes possible. We ranked the ADCs in our study as follows: TMAb mcMMAF > TMAb vcMMAE > Thio-TMAb vcMMAE > Thio-TMAb mcMMAF.
From a CMC development viewpoint with an emphasis on stability, we believe that TMAb mcMMAF is the molecule best suited for production. The mcMMAF also appears to be the preferred linker-toxin for stochastically conjugated TMAb, but it is the most destabilizing for the site-directed conjugated Thio-TMAb. On closer evaluation, the TMAb ADCs gave similar structural results, the main difference being DAR linker-toxin stability. Localization of the linker-toxin to a single specific site appears to be more structurally destabilizing, and the presence of the more hydrophobic mcMMAF seems to exacerbate that effect.
In addition to helping us select a lead candidate, those data also provide information on the lead candidate itself. For instance, our lead candidate can be considered a medium risk with regard to further development. There is evidence that mcMMAF conjugation has a destabilizing effect on protein HOS, making it prone to aggregation. So both process and formulation development will need to take that into account, possibly using surfactants and potentially also other stabilizing excipients from the beginning. Highly concentrated formulation may not be possible for this molecule, and indeed liquid formulations might not be sufficiently stable, so lyophilization needs to be considered.
As is often the case, the data from this study give rise to additional questions. Our results indicate where further analysis with orthogonal techniques is needed to provide a more comprehensive understanding of protein impurities produced as a result of the stress conditions.
Note that a developability risk assessment primarily covers manufacturability and does not necessarily correlate to the in vivo efficacy of these molecules. Hence, developability risk assessments performed for lead selection always need to be evaluated in relation to data from in vivo and in vitro efficacy studies. Although such data are not currently available for these ADCs, the cell-based cytotoxicity assay did reveal that the TMAb mcMMAF molecule was the most potent of the ADC variants. Based on our potency data, a possible explanation is that the stochastic TMAb mcMMAF delivers nearly twice the toxin of its site-directed equivalent. Furthermore, the relative potency of the MMAF conjugates compared with the MMAE conjugates may be lower because of multidrug resistance effects. Free MMAE may be pumped out of cells following cleavage of the linker region, resulting in lower intracellular levels of toxin but potentially a greater bystander effect. We’re planning cell-based assays to further evaluate this and other aspects of the ADC’s mode of action.
Ready in 20 Weeks
Significant research and development is going into producing new ADCs for cancer treatment. The fact that these molecules combine a biologic with a small-molecule drug makes them among the most complex of therapeutics, which can bring significant issues with development for first-in-human studies. Here we have described a fast-track “gene to ADC product” approach using a series of platform technologies to provide a lead candidate rapidly for development. From selection of the gene, an antibody (or antibodies) can be manufactured in laboratory-scale quantities within eight weeks. Similarly, the chemistry for a range of linker-toxin conjugations can be optimized, and developmental material can be produced within four weeks. Finally, a developability risk assessment can provide both a lead candidate and related “prior knowledge” in line with the QbD paradigm within another eight weeks. The overall outcome represents time, cost, and resource savings in the development of an ADC product.
References
1 Beck A, Reichert JM. Antibody–Drug Conjugates: Present and Future. mAbs 6 2014: 15–17.
2 Rehberger B, et al. Accelerating Stable Recombinant Cell Line Development By Targeted Integration. BMC Proc. 7(6) 2013: P111; doi:10.1186/1753-6561-7-S6-P111.
3 Gomez N, et al. Triple Light Chain Antibodies: Factors That Influence Its Formation in Cell Culture. Biotechnol. Bioeng. 105(4) 2010: 748–760; doi:10.1002/bit.22580.
4 Carta G, Jungbauer A. Protein Chromatography: Process Development and Scale-Up. Wiley-VCH: Weinheim, Germany, 2010: 4–5.
5 Chen X, et al. Charge-Based Analysis of Antibodies with Engineered Cysteines. mAbs 1(6) 2009: 563–571.
6 Gerster A, et al. A Simple Method to Determine IgG Light Chain to Heavy Chain Polypeptide Ratios Expressed By CHO cells. Biotechnol. Lett. 38(12) 2016: 2043–2049.
7 Jain N, et al. Current ADC Linker Chemistry. Pharm. Res. 32(11) 2015: 3526–3540; doi:10.1007/s11095-015-1657-7.
8 ICH Q8(R2): Pharmaceutical Development. US Fed. Reg. 71(98) 2009; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf.
9 ICH Q9: Quality Risk Management. US Fed. Reg. 71(106) 2006;www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
Corresponding author Allan Watkinson, PhD, is director of biopharmaceutical development; and William Stimpson, PhD, is CMC sales director at Envigo in Huntingdon, UK; allan.watkinson@envigo. com, william.stimpson@envigo.com. Anja Trapp is a scientist in bioprocess science, and Jadranka Koehn, PhD, is director of cell-line development at Rentschler Biopharma in Laupheim, Germany; anja.trapp@rentschler-biopharma.com, jadranka.koehn@rentschler-biopharma. com. And Colin McKee, PhD, is head of technical services for ADC Biotechnology in St. Asaph, UK; colin.mckee@adcbio.com. Turbo Cell is a Rentschler trademark.