
www.graphicstock.com
In the current global regulatory environment, management and implementation of postapproval CMC changes often can be unpredictable and inefficient. Timelines for change approval can vary from months to years, depending on regional regulatory procedures. Therefore, the challenge in postapproval lifecycle management is to maintain a constant supply of high-quality product while supporting innovation and continual improvement. This was the premise of the CASSS Chemistry, Manufacturing, and Controls (CMC) Strategy Forum held in Gaithersburg, MD, on 20–21 July 2016. The forum explored pathways for operational flexibility in the postapproval phase of the biopharmaceutical product lifecycle and specifically focused on the following topics:
- Current and/or future regulatory pathways or tools that provide global operations flexibility in making postapproval changes (PACs) — and whether these differ for accelerated programs
- Closing the gap between approval timelines (and data requirements) for postlicensure changes between the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) countries and non-ICH countries.
According to the concept paper for the proposed ICH Q12 guideline (Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management), ICH Q12 intends to improve the efficiency of regulatory evaluation, both in review and inspection over the product lifecycle through regulatory dossiers, pharmaceutical quality systems (PQSs), and PAC management plans and protocols (1, 2). In close alignment with ongoing work on the ICH Q12 guidance, this forum highlighted the benefits of clearly defining established conditions (regulatory commitments), with further reliance on the PQS, implementation of postapproval change management protocols (PACMPs), and management of a registration’s level of detail to ensure sufficient manufacturing flexibility (3).
The forum also highlighted discussion of the following questions:
- Will ICH Q12 be transformative?
- What does the biopharmaceutical industry need from ICH Q12?
- What are regulators’ perspectives and opportunities?
- What key principles need to be addressed for ICH Q12 to succeed?
- How will industry and regulators know whether ICH Q12 has succeeded?
| CMC Forum Series |
| The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The Forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS, an International Separation Science Society (formerly the California Separation Science Society), and is supported by the US Food and Drug Administration (FDA). |
The Problem Statement
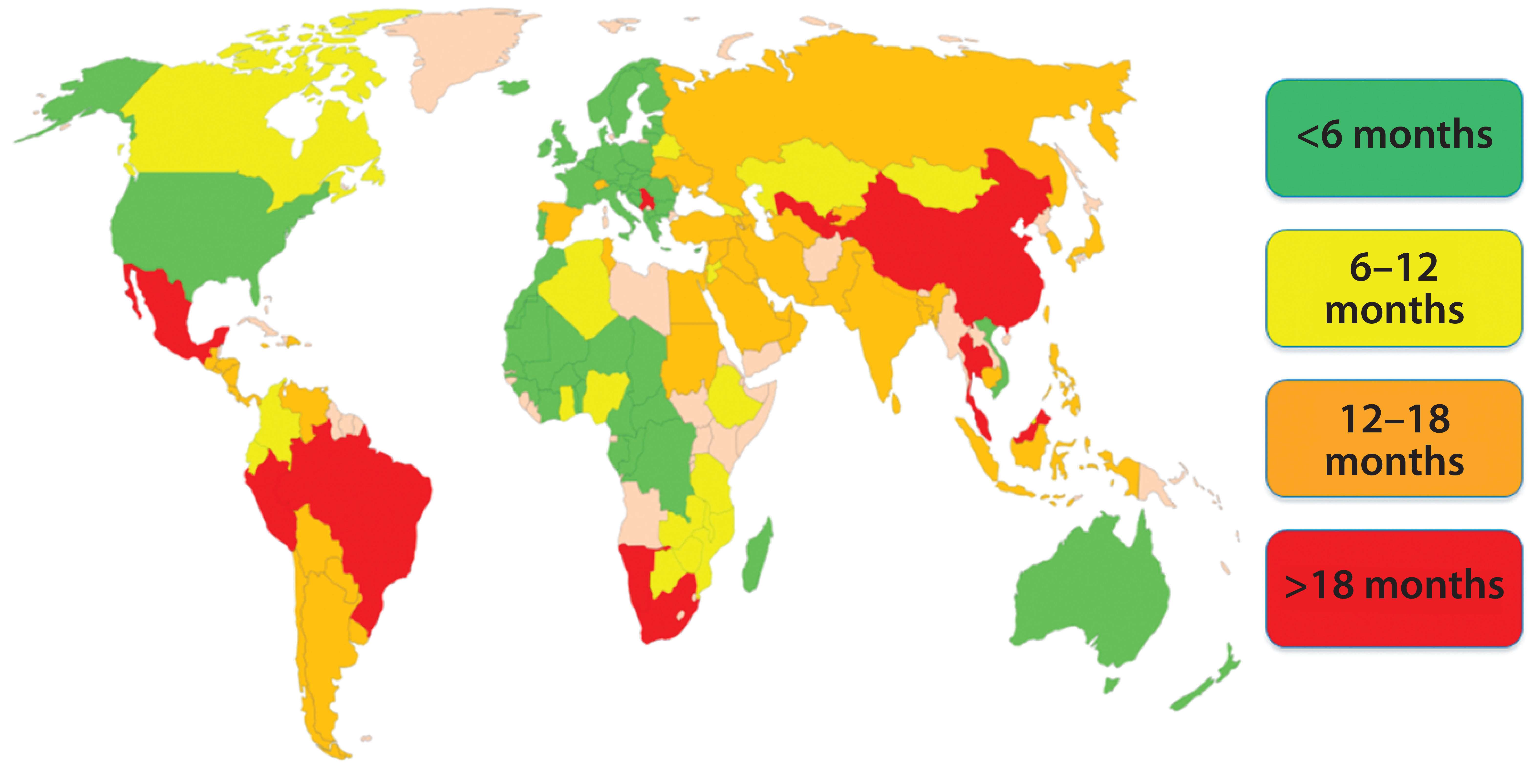
Figure 1: Estimated global approval times for major changes (e.g., new drug-product manufacturing site)
The morning session on Day 1 of the forum was chaired by Yves Aubin (Health Canada) and Joseph Kutza (MedImmune, a member of the Astrazeneca Group). Representing the Biophorum Operations Group (BPOG), Suzanne Murray of Biogen presented “An Industry Perspective: The Complexity of Postapproval CMC Changes and Proposed Regulatory Strategies.” She reiterated that PACs are inevitable and essential, but it is difficult to manage evolving requirements and timelines for every country. BPOG has identified approval timelines around the world (Figure 1); some currently exceed 18 months.
Health authorities — especially those in emerging markets — face a number of constraints: limited resources, processes, and maturity of their regulated manufacturers, as well as both political and legislative considerations. Along with the biopharmaceutical industry, such authorities have identified a number of mitigation strategies to address the challenges of managing postapproval changes, but those are not enough. Harmonization of lifecycle management (LCM) for CMC changes is ideal. It would reduce costs and regulatory burdens on both industry and regulators to ensure that patients have timely access to necessary therapies.
Randall Lapcevich of MedImmune presented “Challenges Posed by Post Approval Changes (PACs) on a Commercial Biologic: A Manufacturing and Supply Chain Perspective.” Such changes present a risk to supply chain agility and a company’s ability to address product demand. Some strategies suggested for mitigating the impact of PACs on both supply chain and manufacturing include addressing foreseeable changes during the original license-application preparations, bundling changes with regulatory filings, and maintaining overall flexibility. However, more could be done to demonstrate equivalence with like-for-like changes. PACs can cause instability in supply and, in extreme cases, cause product shortages. Changes come in all shapes and sizes and can have a dramatic impact on the agility and flexibility of a company’s supply chain.
Gresham Weatherly of AbbVie presented “Managing the Product Lifecycle Continuum Through Postapproval Change Management Plans: An Industry Perspective.” Different, customized dossiers describing the same product are challenging to maintain. Their differences come from a number of factors: e.g., review timelines, stability requirements, PAC regulations, customized dossier details, manufacturers, specifications, country-specific documents, and prespecified supply chains. The United States and European Union have a mechanism by which companies can submit postapproval plans — through comparability protocols in the former and change-management protocols in the latter. However, no defined pathways for PACMPs exist currently in other markets.
Ideally, a marketing authorization would describe a company’s change-management system and include generic protocols describing how subsequent changes would be evaluated. The system and protocols then could be approved globally, so changes would be implemented without requiring health authority approval. Such generic protocols ideally would be submitted with the initial global marketing applications describing how changes should be evaluated so that later improvements could be efficiently filed and implemented.
The session concluded with a panel question and answer session. Panel participants included the speakers mentioned above along with Anthony Mire-Sluis (AstraZeneca), Anthony Ridgway (Health Canada), and Anders Vinther (Sanofi Pasteur). The panel addressed questions on topics such as the practicality of a truly “global” filing, effects of approval timelines for implementation of postapproval CMC changes for a globally filed product, closing the gap between approval timelines and data requirements for postlicensure changes in ICH and nonsignatory countries, and whether ICH Q12 will be enough to address the most critical issues associated with change management for both the biopharmaceutical industry and regulatory authorities.
Q12 Global Status and Impact
The afternoon session on forum Day 1 was chaired by Sally Anliker (Eli Lilly and Company) and Emanuela Lacana (FDA’s Center for Drug Evaluation and Research, CDER). Anthony Ridgway presented “Regulatory Guidance for Product Lifecycle Management: Harmonization Goals, Opportunities and Challenges.” The concept of “established conditions” is a major element of ICH Q12. For existing marketed products, the ICH Q12 Expert Working Group (EWG) is discussing two concepts: default established conditions (those conditions documented in regulation and guidance documents) and negotiated established conditions, (those defined through discussions with the regulatory authority and cemented as part of product approval).
One challenge for Health Canada — and other regulators, probably — is that existing guidance will need to defer to negotiated established conditions and determine how to ensure consistency from company to company, site to site, and product to product. Ridgway says the agency will need to evaluate and address the consequences of a potential increase in PACMPs upon its resources, including potential reductions in revenues from industry submission fees.
Although such protocols are recognized as underused, they do play a significant role in ICH Q12. Multiple-element PACMPs that combine more than one change or more than one product could provide “more bang for the buck,” even when companies are not committed to using a given protocol once it is approved. To encourage broader adoption, it may be possible to capture established conditions, reporting categories, and PACMPs within the ICH Q12 guidance. The EWG is considering a number of additional points for ICH Q12. One point is to address differences between the complexity of products and processes for biologics relative to those of small molecules. Ultimately, ICH Q12 should create a framework that will foster ICH-independent regulatory harmonization and convergence and have value beyond current ICH parties and members.
Wassim Nashabeh (F. Hoffmann-La Roche) presented “Will ICH Q12 Truly Go Beyond Q8/11? Opportunities and Challenges.” Implementation of ICH Q8–Q11 provides opportunities for a more science- and risk-based approach to assessing changes across a product’s lifecycle. Historically, however, the emphasis has been on the development stage of that lifecycle. Opportunities and benefits have not been fully realized or enabled, so the envisioned “operational flexibility” has not been achieved. Thus, the focus needs to shift now to the commercial manufacturing phase, enabling a system that facilitates managing quality and continual improvement throughout the entire product lifecycle with an emphasis on the postapproval phase.
Most pharmaceutical companies operate globally, with manufacturing sites around the word, global product registrations in multiple countries, and global product supply chains. The regulatory environment requires convergence, and a continued fragmented landscape will delay innovation. Currently, global regulatory organizations also have been collaborating to harmonize their expectations and address the challenges of submitting PACs globally. The Asia–Pacific Economic Cooperation (APEC) is working with the World Health Organization (WHO) to focus its efforts on postapproval variations. The Pharmaceutical Inspection Cooperations Scheme (PIC/S) has leveraged common standards and is harmonizing inspection expectations to drive them. WHO has been working on a number of guidelines for PACs and variations of vaccines, prequalified products, and multisourced pharmaceutical products, while other WHO guidelines are under development. All these activities will help integrate a number of ICH Q12 elements around the world.
Established conditions form the core of ICH Q12, having been defined under the current working draft guidance. If this concept is implemented correctly, it will lead to true transformation of LCM and submission transparency. Whether or not a given parameter is identified as an established condition, the manufacturing process remains the same based on patient benefit/risk linked to product/process understanding. The only difference would be the extent of review by a regulatory agency. For continued success, the industry must strive toward harmonized international regulatory policy with reduced regional or local requirements by implementing the spirit of ICH and expanding adoption to all countries.
Mahesh Ramanadham (FDA CDER) presented “ICH Q12: A Much Needed Culture Shift.” If ICH Q12 is driving opportunities for companies to prospectively manage future changes more strategically, the desired state for those companies will be to manage most manufacturing changes effectively under their PQSs without need for regulatory approval before implementation. ICH Q12 highlights two enablers: a robust product/process understanding, and an effective PQS. An effective quality system will drive each company toward a positive quality culture and provide confidence that most changes can be managed solely under that PQS.
Quality systems are expected to manage both established conditions and nonestablished conditions. ICH Q12 will change the focus from the original license application (currently only a milestone) to the entire product lifecycle. This includes not only development, but also the dossier and management of established conditions, PACs and change protocols, and regulatory commitments. It will require all elements of the product lifecycle to work together and make a culture shift necessary. That change in culture should allow for enhanced transparency and trust between companies and regulators, focus time and efforts on higher-risk issues, and shift more postapproval LCM back to the industry (and the pharmaceutical quality system).
Yasuhiro Kishioka from Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) presented “Postapproval Changes in Japan” with an overview of the regulatory framework there. Currently the company recognizes two types of changes under its partial change applications and minor change notifications. When the latter was introduced, harmonization among ICH regions was considered, but the PAC reporting categories is not harmonized across the ICH regions.
The concept of established conditions is critical to development of a transparent, flexible, and effective regulatory framework under ICH Q12. In Japan, approved matters captured in application forms are very similar to the concept. PACs that do not affect approved matters are managed within a PQS. In some sense, an application form becomes the basis for established conditions used to determine filing strategies for PACs in Japan. Because this approach is unique to Japan, it presents a challenge for the ICH Q12 EWG.
A question-and-answer panel followed the afternoon presentations, with Patrick Swann (Biogen) joining the afternoon presenters. Questions asked focused on a broad range of topics, including how to define core values for quality culture, harmonizing the initial marketing authorization application across different regions to identify a sponsor’s established conditions, defining success for ICH Q12 and how it will address transparency relating to scientific risk and trust, and the ramifications possible with a loss of trust between industry and the regulatory agencies. The main themes of the afternoon centered on success factors for ICH Q12 (including harmonized definitions for established conditions and an approach for PACs), challenges for non-ICH countries, the importance of trust between industry and regulators, and the need for companies to manage and maintain established conditions.
The Reality of ICH Q12 Concepts
Julia Edwards (Biogen), Michelle Frazier (AbbVie), and Ingrid Markovic (FDA CDER) chaired the morning session on the forum’s second day. Titled “Changing Lifecycle Management in Practice: The Reality of ICH Q12 Concepts,” this session advanced the discussion of effective PAC management by overviewing case studies of how ICH Q12 ideas could be implemented by the biopharmaceutical industry. Practical examples provided by speakers and attendees actively participating in panel discussion elucidated revolutionary aspects of ICH Q12 and provided useful feedback for the EWG to consider during upcoming ICH Q12 meetings. The session included three presentations and an active panel session that highlighted key points regarding established conditions of analytical methods and considerations for outlining what is an established condition within a regulatory dossier.
Bob Iser (acting director of FDA CDER’s Office of Process and Facilities), presented on behalf of his fellow FDA EWG team members and contributors Ashley Boam, Ingrid Markovic, and Mahesh Ramanadham. His talk, “FDA Perspectives on Established Conditions and ICH Q12,” highlighted opportunities and challenges of Q12, the only ICH guideline that includes both regulatory and technical considerations. Opportunities include harmonization, flexibility, encouragement of continual improvement, and general sharing of knowledge to allow better decisions to be made for all products. Challenges include convergence across regions and implementation of Q12 concepts for legacy products. The presentation focused on established conditions and included many thoughtful considerations for linking them to a control strategy and change management, including understanding the criticality and risk associated with certain conditions (4).
James Sesic (Amgen) presented “Complexities of CMC Change Management and Q12 Opportunities,” reviewing the present realities of advancing biological changes and the complexities of lifecycle management. He noted a need to implement hundreds of manufacturing and quality control changes per year. Implementing those could take three to five years in some countries, complicating the tracking of such changes. The industry can use effective strategies to manage that complexity and anticipate both the opportunities and challenges of ICH Q12 (such as optimizing the format of Module 3 for global submissions). Additional opportunities for industry include the possibility of creating consistent opportunities for non-ICH countries to help implement changes globally.
Kim Wolfram (Biogen) presented “20/20 Vision: The Future of ICH Q12 in Practice,” considering what it would be like for patients after ICH Q12 has been implemented (in the year 2020). She showed why concepts outlined by ICH Q12 are not simply “nice to have” but rather “must-have” for products of the future. Through effective integration of product knowledge (knowledge space), robust risk assessments (identifying criticality), and adaptive controls and/or scientific models, a company could expect less uncertainty and propose fewer established conditions.
Opportunities to leverage output-based established conditions will ensure that ICH Q12 is transformative and best able to meet the demands of future technology and enhanced knowledge. That is predicated on the fact that robustness will be built into each manufacturing process, so each process will have the ability to withstand unforeseen events. Open questions for this approach are related to the level of detail required in a dossier for regulatory submission and how to identify content for both established conditions and those not considered to be such.
Morning Discussion Points
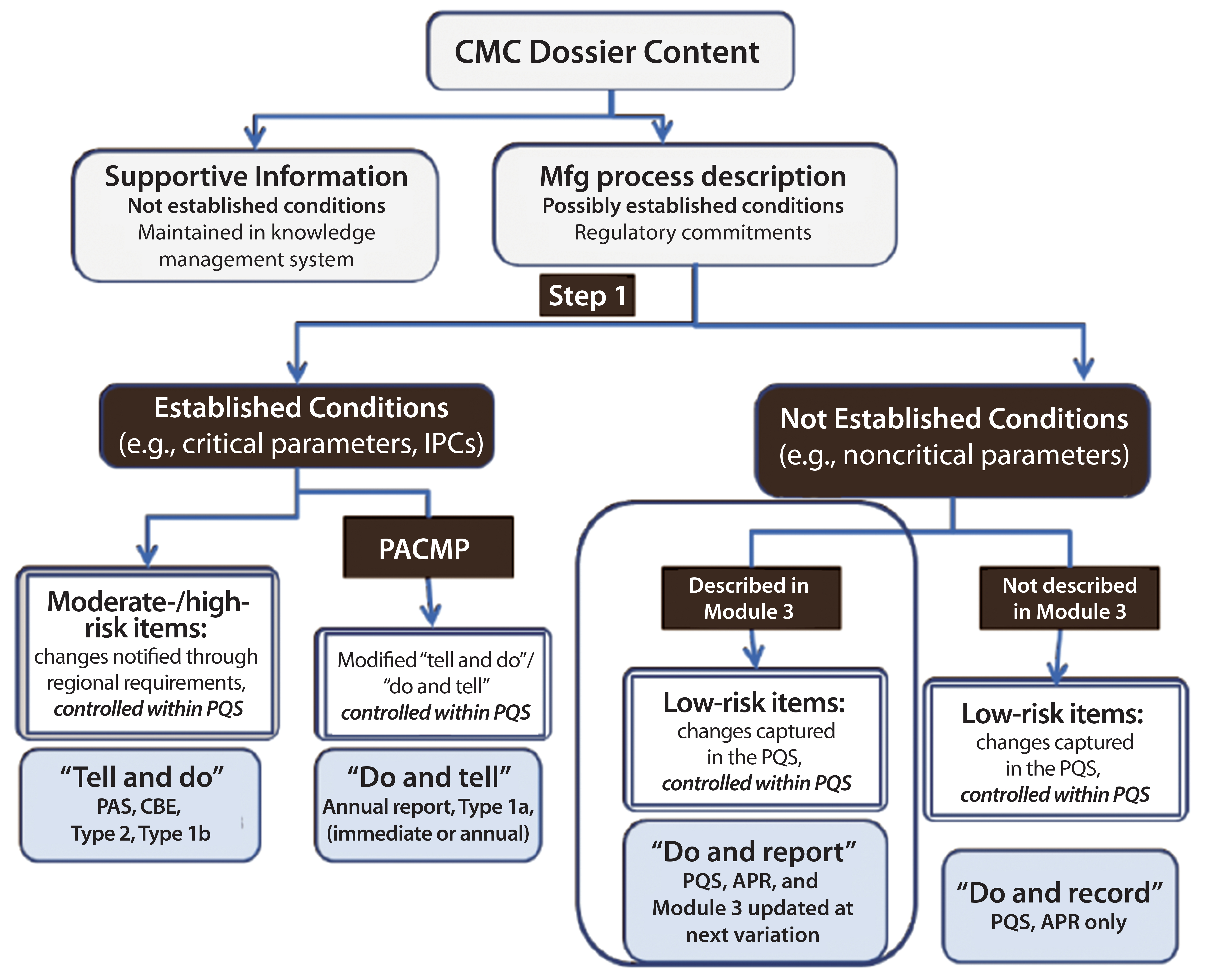
Figure 2: Contents of a chemistry, manufacturing, and controls dossier; APR = annual product review; CBE = changes being edited; IPCs = in-process controls; PACMP = postapproval change management protocol; PAS = prior approval supplement; PQS = pharmaceutical quality system. Source: Robert J-L, Cook G. European Industry Workshop on Lifecycle Management. Joint Biologics Working Party/Quality Working Party/Good Manufacturing and Distributing Practice Inspectors Working Group.
A question-and-answer panel included Sally Anliker, Ashley Boam (FDA CDER), Jennifer Mercer (Genentech, a member of the Roche Group), James Sesic, and Kim Wolfram. The main themes of the morning fall into three topics: practical considerations for established conditions (including for legacy products), the PQS and managing established conditions over a product’s lifecycle, and interagency communication related to LCM for FDA product reviewers and inspectors. A general theme throughout all sessions was to ensure that these concepts get discussed with non-ICH countries. Such countries pose the most challenges regarding change management, and although they may not follow ICH Q12 once it is implemented, they may be encouraged to begin adopting some of its principles.
Established Conditions — Practical Considerations: Established conditions were mentioned often, not only as a foundational element of ICH Q12, but also as having the most potential to ensure that the guidance reaches its goals. In particular, there was general consensus that output-based established conditions will provide the most benefits and are not driven merely by a desire to do less testing; in many ways, the approach would increase testing. Forum participants generally accepted the current definition of established conditions and engaged in deep and rich discussions related to how and where established conditions will be outlined within regulatory dossiers, as well as how to ensure that LCM of established conditions does not erode the benefits of ICH Q12.
That document and specifically the assignment of output-based established conditions, is predicated on the effective use of robust risk assessment and expanding product knowledge. The question that a drug sponsor must ask is whether it has sufficient knowledge space of its product and process to understand what factors impact their CQAs, which will enable output-based established conditions. ICH Q12 is intended to build in that flexibility to align with the increase of product knowledge over the lifecycle of each drug product. However, attendees noted that there may not be the same level of knowledge for each product and there is no “one size fits all” solution. Several examples illustrated the use of output-based established conditions, which leverage risk assessment and product knowledge. Such conditions are a necessary enabler for the use of enhanced technologies, such as adaptive process controls, but they do require robust product and process knowledge to justify their use. Forum participants acknowledged that it is possible to remove established conditions throughout a product’s lifecycle through continuous process verification by enabling increased product and process knowledge. Such knowledge could be linked to a lifecycle management plan (LCMP) and comparability protocols to enable LCM of established conditions. Attendees generally agreed that removing established conditions does require a filing and sufficient justification.
Key consensus among industry participants regarded ensuring clarity and consistency across regions — rather than centering established conditions on increased flexibility in global change management. Transparency is important to accomplishing this. Established conditions should not introduce new layers of complexity and controls, but rather introduce clarity and consistency that will benefit regions without clear guidelines in place. Forum participants acknowledged that no one wants to change regulations — and in some cases, that cannot happen — but that everyone wants clearer guidance.
FDA representatives noted that listing established conditions is not required in current applications, and if sponsors propose to include such a list in forthcoming dossiers, they should provide advance notice to the agency. In some cases (e.g. breakthrough therapies), it was noted that up-front alignment and discussion of proposed established conditions would be needed to ensure that products be made available to patients with unmet medical needs while maintaining a feasible LCM approach for established conditions. FDA representatives noted that once ICH Q12 is final, the agency will need to train reviewers on how to review submissions that include established conditions appropriately, and that doing so will take time.
| What Is an LCMP? |
| As defined by the Joint BWP/QWP/GMDP IWG European Workshop on Lifecycle Management in the Application of ICH Q12 Tools and Enablers: Lifecycle Strategy, a LCMP provides a single, holistic framework giving a big-picture view for regulators that links realization of a quality target product profile (QTPP) through the control strategy; defines both established and nonestablished conditions; includes a plan for postapproval continual improvement; facilitates more commonality in review across ICH regions; promotes achievement of a single approved filing on a science- and risk-based oversight, and positively supports innovation and continual improvement. |
Analytical Methods: As scientific technologies advance in the field of analytical methods — and most PACs are intended to improve such methodologies — the need for clear, predictable, harmonized, and consistent established conditions for test methods becomes exceedingly important. Forum participants acknowledged that they may not have all the detailed technical knowledge needed to discuss this topic fully, but they generally believed that any method change affecting specifications should be considered to be an established condition. There was also general consensus that method-performance characteristics should be established conditions and that method-validation information is supportive only and thus would not be considered an established condition (although validation criteria should be met to ensure adequate method performance).

Table 1: Making changes to analytical methods — an example
Kim Wolfram (Biogen) offered an example focusing on a change related to size-exclusion chromatography (Table 1) as it relates to key method performance characteristics. In this example, the established conditions are the basis of separation and detection, system-suitability criteria, and assay and sample acceptance criteria. The nonestablished conditions would be the column, solutions, or quantities used. That would facilitate a change from high-performance liquid chromatography (HPLC) to ultraperformance liquid chromatography (UPLC) that is managed solely within the PQS and not reported to any regulatory agency. However, this example would require validation in accordance with available ICH guidance.
One attendee raised a concern over how to address method changes when in-country verification testing is required. Another suggested leveraging the available method as long as it was proven to be equivalent to the updated method. That could pose challenges for drug sponsors to ensure that their methods are kept updated and will not produce unexpected results. That challenge relates back to a need to elicit discussion with non-ICH countries about this topic.
Ultimately, the responsibility of a product sponsor is to use quality risk management to determine whether the potential exists to compromise drug quality and whether risks are introduced. If the risk-assessment output confirms an absence of such impact to product quality, then a given change could be managed by the PQS.
Legacy Products: One conundrum in development of ICH Q12 is how to best meet the needs of legacy products, with the unique and formidable challenges that such products introduce. Use of available ICH guidance has failed to address those challenges, and that has led to a surge in both the number of postapproval filings and the amount of content included in regulatory dossiers. These challenges were outlined nicely in the problem statement segment at the beginning of this forum.
The goal of ICH Q12 is “to reduce unnecessary cost and time burdens on industry and regulators, while assuring patients reliably have access to high quality therapies” — and that will need to include legacy products, which are the primary therapies on the market today. Two types of practical benefits could be realized: providing guidance for changes for which technical requirements are addressed by existing ICH guidelines, and using broad PACMPs for certain changes across multiple products and/or manufacturing sites. Those two areas are expected to present challenges in global harmonization, but the EWG commitment to address them is encouraging. Several global regulatory agencies there are committed to trying to implement PACMP-type pathways where none currently exist.
Pharmaceutical Quality Systems and ICH Q12: Forum speakers introduced the concept of an effective PQS, highlighting that as a necessary and enabling aspect of ICH Q12. Themes of trust, change management, and risk assessments emerged in relation to the topic. FDA-CDER’s Iser encouraged the forum to reread ICH Q10 (1), which will be used to identify and evaluate whether sponsors have an effective PQS. Q12 is intended to build on Q10 by providing further clarification and consistency. It will aid in ensuring trust and confidence in a sponsor’s PQS such that global regulatory agencies fully understand how risk assessments and efficient use of product and process knowledge are used to drive control strategies. Key PQS concepts that are likely to be mentioned in Q12 include effective change management, corrective and preventive action (CAPA), process performance and product quality monitoring system, management review, outsourcing and PQSs, benefits of an effective PQS, and use of knowledge management in LCM.
Lifecycle Management Plan Strategy: The benefits of having a centralized place to capture all information related to established conditions have been acknowledged often throughout the ICH Q12 discussions. One such place could be an LCM plan/strategy document, which is a key concept in Q12. Discussions continue over what information should be built into such a document and what would be considered binding. Attendees expressed some general concern that if reviewers disagree with the information in the plan/strategy document, that could compromise a product’s approval overall.
Communication Between Product Reviewers and Inspectors: One topic that emerged from the morning discussion was a need for increased communication between inspectors and product reviewers. An example one forum participant described was detailed drug-product media filling information requested by FDA microbiology reviewers as part of Module 3 information — items that could and should be viewed commonly upon inspection. The agency is using an integrated inspection/review model that promotes open communication (and is working to improve that further), but forum participants noted that several areas could benefit from increased discussion to help streamline information requested for Module 3.
Managing Postapproval Changes in Non-ICH Countries
Michael Abernathy (Amgen) and Michelle Frazier (AbbVie) chaired the afternoon session entitled “To Infinity and Beyond . . . Managing the Myriad Postapproval Changes in Non-ICH Countries.” This session focused on blue-sky PAC management concepts such as the feasibility of having a single, core marketing application dossier. Q12 holds great promise for ICH countries, where a single set of registered conditions and postapproval change-management protocols will speed implementation and ease the burden of change control (albeit after full alignment with legalities in all ICH countries is achieved). The ability to leverage its proposed benefits can improve lifecycle consistency for non-ICH regions as well by increasing availability and continuous improvement of existing therapies. This panel included a member of ANVISA (the National Sanitary Surveillance Agency of Brazil), which became a full ICH member in November 2016. The ANVISA perspectives and commitment to this topic show great promise for advancing improvements to postapproval change management, although resourcing constraints continue.
Vinther of Sanofi Pasteur presented “Improving Postapproval Change Processes As a Way to Ensure Technical Innovation and Drug Product Availability.” He noted that the promises of ICH Q10 and process analytical technology have not been realized fully and in fact are moving at a slower pace than many had hoped. Regulatory processes vary among different countries in their requirements and timelines, which presents a logistical challenge for companies managing several product versions simultaneously. That in turn increases risk to product availability. Increased dialogue between industry and regulators is needed to facilitate more innovation and improve drug-product availability. A proposed solution includes reduced regulatory filing submission burdens and establishing a strong science- and risk-based approach to PACs. Anders also touched on the benefits of having an LCM plan built into your initial dossier and how that could facilitate PACs through transparency and mutual understanding.
Robert Laughner (MedImmune) presented “The Device Side of Combination Products: Technical and Regulatory Challenges in Life Cycle Management.” He focused on the fact that change management of medical devices does apply to combination products and thus should not be overlooked. In fact, current FDA guidances state that changes to drug-product container–closure systems require prior-approval supplements (PASs) depending on what a given change actually is — but that the requirement could be interpreted to cover nearly any change. Under such an interpretation, management of container–closure improvements and updates after product approval could become problematic. Comparability protocols should be used often to help provide regulatory relief.
Finally, Laughner highlighted a gap in combination-product discussions for ICH Q12. He noted that the FDA was set to release a draft guidance on postapproval modifications to combination products by the end of 2016 (but as of July 2017, it has not been published yet) and that the Combination Products Coalition was working on proposed revisions already. Mercer of Genentech gave a talk titled, “The Potential Benefits and Challenges of ICH Q12 for Managing Global Changes.” She included a few case studies illustrating management of seemingly simple postapproval changes.
One example was a minor change to a test method, which is considered by some countries to be critical and require prior approval and by others to be minor and not require reporting. Final approvals for such a change took up to 20 months. Other experience has indicated that global implementation of a change could take up to five years or even longer if it leads to increased reporting and requests for more details. Mercer noted throughout her talk that country-specific established conditions may be unavoidable and that management of changing them will be difficult. As a result, Q12 may be truly beneficial only if performance-/output-based established conditions are enabled.
Afternoon Discussion Points
A panel discussion concluded the forum and touched on themes that had come up both days. Panelists included Iser, Laughner, Mercer, Murray, and Vinther. Here it was reiterated that Q12 is intended to promote innovation and trust in quality systems and that building that trust requires improving PQSs to be more robust. Additionally, the general consensus was that Q12s principles may be easier to implement for new applications than for legacy products and that regulatory review of applications are not useful if they don’t add value.
Postapproval Changes in Non-ICH Countries: WHO guidance has been used by some companies considering postapproval changes beyond the ICH regions. An example is WHO Technical Report Series 943 (Annex 6) specific to PAC management for approved vaccines. Panelists indicated that the WHO document is an excellent source that is used increasingly. Its strength is that it is prescriptive, which was viewed as the primary reason for its increasing use and adherence.
Therefore, forum participants considered whether the WHO document could be viewed as complementary to ICH Q12. This idea was specifically noted in discussions of the standardization process advocated within the WHO framework. One attendee noted during the question-and-answer period that the WHO guidance is voluntary and the current FDA framework does not require compliance with it. It can be used as a reference within other documents, but it is generally understood that the guidance will be used primarily for non-ICH regions and that harmonization between ICH and WHO could be improved.
Participants also noted that non-ICH countries are working to meet the needs of an increased number of supplements while balancing (in some areas) a reduction in resources. Even if review times are reduced, that does not address the issue of backlogs. To combine the reduction of review timelines with a reduction in the time it takes from when an agency receives a submission and begins its review is an issue that exists both within and outside ICH regions. The problem was noted to be more of a challenge to non-ICH countries because of resource constraints.
Combination Products: Can We Learn from Device Practices? An increasing number of biologics are combined with medical devices in combination products. Changes for such products are introduced to improve device performance, increase product safety and/or usability, improve drug manufacturability or production yields, and add new device features or functionalities — or they may be based on supplier changes, material/component improvements or discontinuations, and product complaints or CAPAs. Currently no postmarket submission requirements are specific to combination products, so changes rely on available regulations or guidance specific either to a drug or device (constituent part).
The International Pharmaceutical Aerosol Consortium on Regulation and Science (IPAC-RS) Device Working Group is making an effort to create a proposed framework for addressing combination-product design changes and associated regulatory submissions. This framework is built on the drug paradigm (e.g., ICH guidelines) rather than device paradigms (e.g., 21 CFR 820.30 and ISO 13485). The FDA also is working on a draft guidance for postapproval modification of combination products. And the International Standards Organization (ISO) has initiated development of new standards related to LCM of combination products.
Use of LCM for combination products poses many challenges. However, existing device frameworks can be used as supportive information in development of drug regulations and guidance. A lack of definition related to the exact submission type for PACs will continue to be ambiguous, but many cross-industry collaborations are dedicated to improving LCM for combination products. Because regulations vary across regions, the vast majority of delivery-system products are filed with minimal guidance of what defines an established condition. In addition, confusion reigns over whether PAC reporting is required for such products and how to assign reporting categories to them.
CASE Study — Vaccines: It is well understood that biologic drug products are more complex than small-molecule products. That complexity extends beyond characterization and into lifecycle management. A compelling example for this comes with vaccines. If such a product consists of not one but eight antigens (or drug substances), then both its sponsor and regulators must consider changes that are not specific to just a single manufacturing process, but to multiple processes. The complexity increases further when a number of drug-substance and drug-product manufacturing sites are involved. Although change is a natural and necessary part of innovation, it is increasingly burdensome to consider how to introduce it into such a complex matrix. The current PAC management environment leads to drug shortages and highlights questions related to control. “How can regulators and industry work together to grapple with challenges such as this?” forum participants asked. “Do they limit innovation?”
Common Objectives — Elements of Successful Change Management: As industry and regulators await publication of ICH Q12, many industry best practices related to LCM of complex products could be implemented today. Such solutions also could help address Q12’s perceived shortcomings. Sanofi Pasteur’s Vinther and Genentech’s Mercer outlined some suggestions. In this forum, the change-management dialogue that is being generated by ICH Q12 was overwhelmingly viewed as a positive step. Clearly regulators and industry share similar objectives but are approaching the issue from different directions. So collaboration and communication must increase. Vinther stated that the current system is not sustainable because it hinders innovation and causes drug shortages.
The biopharmaceutical industry pursues changes for a number of different reasons: There are necessary changes, voluntary changes, and those pursued to maintain good manufacturing practice (GMP) compliance. Vinther suggested that industry should clearly articulate how expedited and harmonized PACMPs enhance redundancy, improve process and product controls, increase cost-efficiency, reduce lead times, and add value through introduction of new technologies. On receiving such clear articulation — which could be accomplished using LCMPs — regulatory agencies could evaluate risks and benefits of each proposed change without having to guess why a sponsor is advancing it. And they could better understand a sponsor’s rationale for prioritizing that change.
Vinther also recommended specific and harmonized validation and comparability studies for new manufacturing concepts (e.g., single-use systems, continuous manufacturing, modernization of aging facilities, and technologies to manufacture advanced therapies). If the industry can reuse comparability protocols, then their rigor and testing plans could be evaluated before advancing a change. Robustness of protocols and associated data-collection plans would be supported by a sponsor’s quality risk-management program.
Vinther suggested that the industry can advance technologies more effectively if companies speak up and work together to drastically improve the pace of innovation; shift their dialog to be more scientific; articulate the value of technology innovation and more operational leadership involvement; and articulate the need to reduce regulatory burdens.
One World, One Regulatory Standard
As the biopharmaceutical industry grows and more biotechnology-derived and biologic products receive market authorization, the complexity of LCM for CMC changes becomes ever more apparent. Innovation is proceeding, and new technologies are being developed faster than they can be implemented practically. Converging and harmonizing regulations and guidelines (wherever possible) with regard to the development and review of regulatory submissions — and in manufacturing, testing, and release of products — will provide the industry with more resources to implement new ideas more rapidly for the benefit of more patients. “Convergence and harmonization” was the theme of CASSS’s 2017 Well Characterized Biotechnology Products conference this past winter in Washington, DC. This topic came at an ideal time while industry and regulators are advancing with ICH Q12 and beyond.
| CMC Strategy Forum North America Program Committee |
| Siddharth Advant (Celgene Corporation), Yves Aubin (Health Canada), John Bishop (CBER, FDA), Barry Cherney (Amgen Inc.), JR Dobbins (Eli Lilly and Company), Julia Edwards (Allergan), Sarah Kennett (CDER, FDA), Joseph Kutza (MedImmune, a member of the AstraZeneca Group), Kimberly May (Merck & Co., Inc.), Anthony Mire-Sluis (AstraZeneca), Stefanie Pluschkell (Pfizer, Inc.), Nadine Ritter (Global Biotech Experts, LLC), Dieter Schmalzing (Genentech, a member of the Roche Group), Timothy Schofield (GlaxoSmithKline), Zahra Shahrokh (ZDev Consulting), Jeffrey Staecker (BioPhia Consulting, Inc.), Andrew Weiskopf (Biogen), and Marcel Zocher (Bristol-Myers Squibb Company) |
| Disclaimer |
| The views and opinions expressed by individuals do not represent the views and opinions of their affiliated organizations (e.g., company or health authority). The version ICH Q12 document has been updated since July 2016. All statements regarding its content should be subject to review and confirmation of the current available version of the document. |
References
1 ICH Q10: Pharmaceutical Quality System. US Fed. Reg. 74(66) 2009: 15990–15991; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf.
2 EMA/CHMP/CVMP/QWP/ 586330/2010. Questions and Answers on Post Approval Change Management Protocols. EMA Committee for Medicinal Products for Human Use 30 March 2012; www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/04/WC500125400.pdf.
3 CDER/CBER. Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products: Draft Guidance for Industry. US Food and Drug Administration: Rockville, MD, May 2015; www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm448638.pdf.
4 ICH Q10: Pharmaceutical Quality System. US Fed. Reg. 74(66) 2009: 15990–15991; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf).
Julia Edwards (currently executive director at Allergan) was at the time of this forum director at Biogen. Corresponding author Joseph Kutza is director and group manager at AstraZeneca (kutzaj@ medimmune.com). Michelle Frazier (currently vice president of regulatory affairs at Cohherus Biosciences) was at the time of the forum senior director of CMC regulatory affairs at AbbVie, Inc. Ingrid Markovic is senior science advisor for CMC at the US FDA’s Center for Biologics Evaluation and Research; Emanuela Lacana is associate director of biosimilars and biologics policy at the US FDA’s Center for Drug Evaluation and Research. Demetra Macheras is director of regulatory policy and intelligence at AbbVie, Inc. And Kim Wolfram is associate director of regulatory affairs CMC at Biogen.