STOCK.ADOBE.COM
A biosimilar biotherapeutic product is similar (but not identical) in terms of quality, safety, and efficacy to an already licensed reference product. Unlike generic small molecules, it is difficult to standardize such inherently complex products based on complicated manufacturing processes. Table 1 describes the main differences between biosimilar and generic drug molecules.

Table 1: Major difference between biosimilars and generic drugs
The global biosimilar market is growing rapidly as patents on blockbuster biologic drugs expire (Table 2) and other healthcare sectors focus on reduction of costs. Biologics are among the highest-cost treatments on the global market today, which implies the need for low-cost alternatives. In emerging markets, biosimilars already offer more affordable prices, which are not only attractive, but indispensable to economies where expensive treatments are not financially feasible (1). Interchangeability of biosimilars could have a big impact on drug budgets around the world. However, concerns remain about the effect that could have on patients in terms of safety and efficacy.
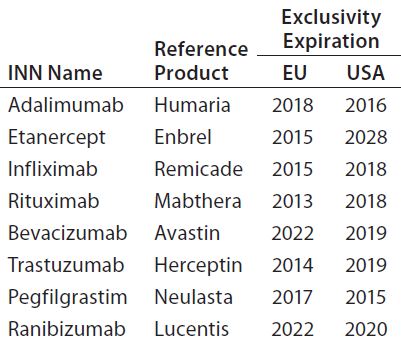
Table 2: Patent status of some innovator biologics (3)
Developing and manufacturing biosimilars is challenging, so well-established biopharmaceutical companies are investing in these important medicines. As Table 3 shows, Europe is leading the way. The United States approved its fourth biosimilar in September 2016, compared with 15 products — marketed under 26 distinct brands — already approved by the European Medicines Agency (2, 3). The market will continue to grow as bestselling biologics come off patent in coming years (4). Hundreds of companies worldwide are developing biosimilars to target diverse markets.
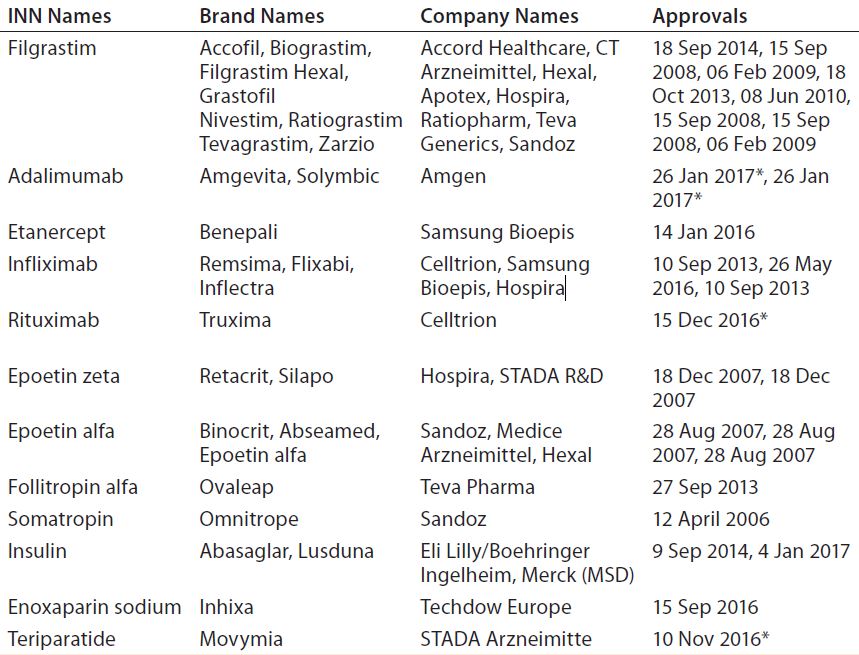
Table 3A: Biosimilars approved on the European market (5); * CHMP positive opinion

Table 3B: Biosimilars approved on the US market (5)
Regulatory Framework of Biosimilars
Table 4 presents biosimilar regulatory pathways for Europe and the United States. The European Union (EU) pioneered development of regulatory requirements for biosimilars in 2005. The EMA also was the first regulatory agency to authorize biosimilars for market. Europe’s extensive experience gained with licensed biosimilars has led to robust regulatory processing by the EMA, with a recent guideline revision adopted by the Committee for Human Medicinal Products (CHMP) on October 2014. In 2015, the first US biosimilar — Zarxio (filgrastim) from Sandoz — encouraged development of biosimilars for that country as well. The US Food and Drug Administration (FDA) released its final biosimilar guideline on 28 April 2015.

Table 4: Regulatory framework in Europe and the United States — differences between European Medicines Agency (EMA) and Food and Drug Administration (FDA) pathways; areas of overlap are tinted.
Biosimilar Nomenclature: The complex nature of biological molecules requires specific nomenclature guidelines. Naming biosimilars has further increased this complexity, and to date, several different and inconsistent conventions have been applied around the world (6). Often, biosimilar and reference products may share the same name. Together with naming inconsistencies, that has led to concern over the strength of the World Health Organization’s International Nonproprietary Name (INN) system currently in place.
The FDA defined how biologicals should be named in a January 2017 guidance (7), which states that each biosimilar must have a proper name made up of a core name hyphenated to a four-letter suffix representing the developer. For example, Adalimumab-atto (Amjevita) is a biosimilar of AbbVie’s Humira drug.
Those FDA-designated suffixes should prevent inadvertent substitution of products. The agency states that these products thus will be distinguishable so that only products that have been approved as interchangeable biologicals (biosimilars) for a particular indication will replace innovator treatments for that indication. This is intended to prevent accidental alternation between different biological products that share the same core name.
Biosimilars are receiving approval in Europe before receiving it in the United States. However, no naming convention has been established in Europe to date. There, biosimilars share the same INN with innovator products, which can create confusion among healthcare professionals.
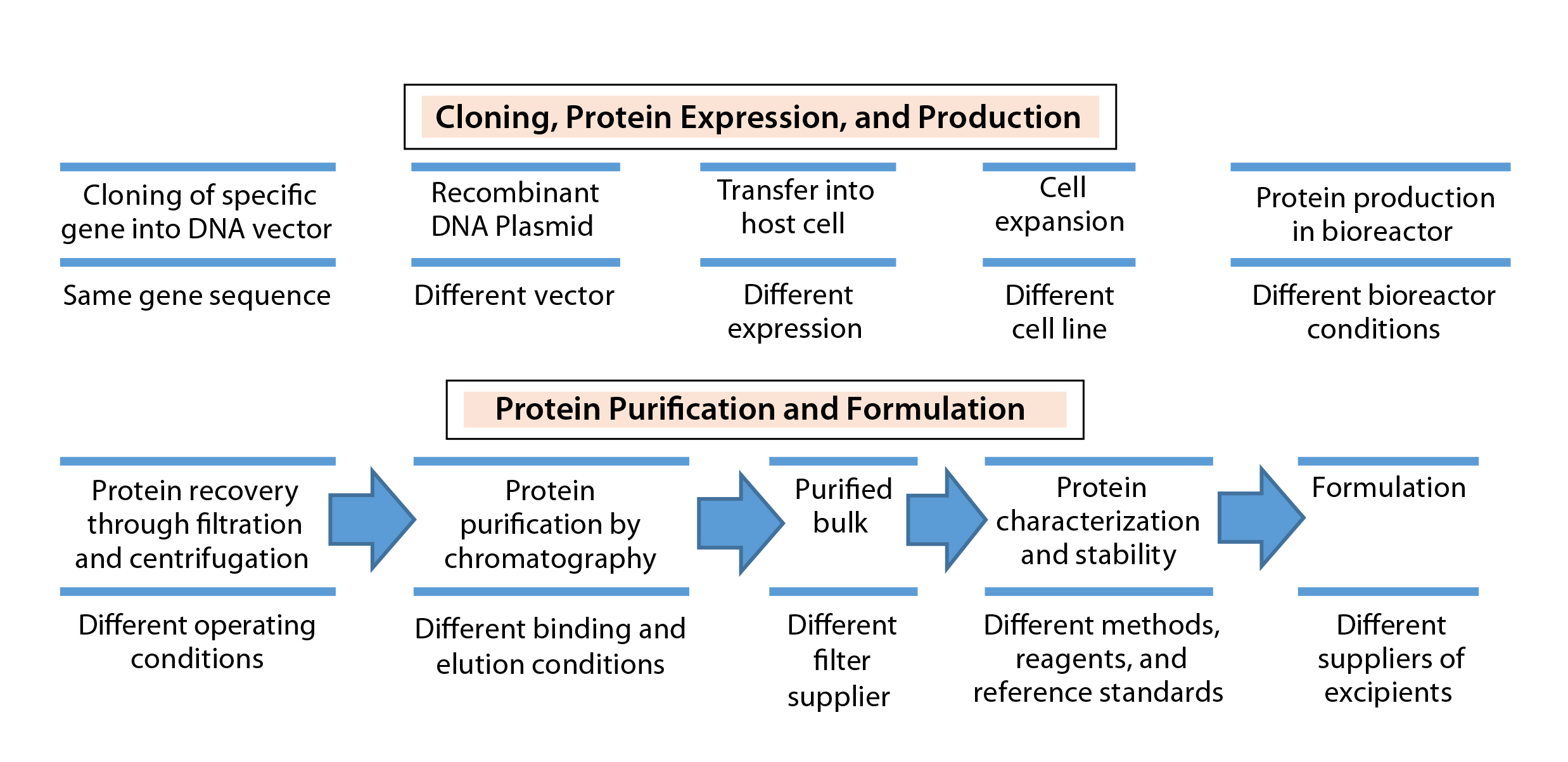
Figure 1: Biosimilar production: source of variation between manufacturers (24)
Biosimilar Development and Manufacturing
Therapeutic proteins derived through recombinant DNA technology (Figure 1) can vary in their primary amino acid sequence or through modifications made to their amino acid chains (e.g., glycosylation, PEGylation, or addition of other side chains to form a secondary structure) and in their higher-order structure (e.g., folding to form a tertiary structure, more complex interactions to form a quaternary structure). Proprietary biomanufacturing processes and environmental conditions used in development of innovator products usually are difficult for biosimilar manufacturers to replicate. So biosimilars are highly unlikely to be completely identical to comparator products.
First, the DNA sequence that encodes a desired biosimilar is identified, isolated, inserted into a vector, and incorporated into the genome of a suitable host cell (e.g., bacterium or mammalian cell). Bacterial host cells are inexpensive and easy to grow, and they generate high product yields. But they cannot produce large, complex proteins such as MAbs. By contrast, mammalian cells do so, but they are more sensitive and costly, and they generate relatively low product yields. A master cell bank with identical cells that produce a desired protein is established through cell screening and selection. That bank is used to culture additional cells at increasing scale under strictly defined conditions that optimize protein production.
In downstream processing, undesired proteins and other impurities are removed from culture supernatant. Harvested protein is analyzed for uniformity in its three-dimensional structure and potency using a number of analytical methods, including physicochemical and biological tests. Finally, the purified drug substance is formulated with added excipients (e.g., antioxidants, osmotic agents, and buffers), filled into containers and external packaging, then stored and shipped under appropriate environmental conditions.
Biosimilar use of different expression systems from those producing reference drugs can change a protein’s posttranslational modifications (e.g., glycosylation profile), which in turn can affect product safety or effectiveness (8). Modification of any steps in biomanufacturing (e.g., use of a different vector to create host cells, systems for cell screening and selection to establish the master cell bank, culture media, methods for production or purification, and excipients) can alter the effectiveness and safety of a product. Thus, biosimilar manufacturers must assess the effects of such changes using appropriate analytical methods, functional assays, and animal and clinical studies to ensure that such changes do not adversely affect the identity, quality, purity, potency, safety, or effectiveness of their products. That also is a question addressed by regulatory agencies when they evaluate biosimilars for approval.
Reference Standard Selection: The EMA has clear guidelines on use of reference standards for similar biological medicinal products (9). To facilitate the global development of biosimilars and prevent unnecessary repetition of clinical trials, it may be possible for an applicant to compare its biosimilar in certain clinical studies and in vivo nonclinical animal studies with a reference product that is not authorized in the European Economic Area (EEA), but that comparator should be authorized by a regulatory authority with similar scientific and regulatory standards (e.g., signatories to the International Council on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use, ICH).
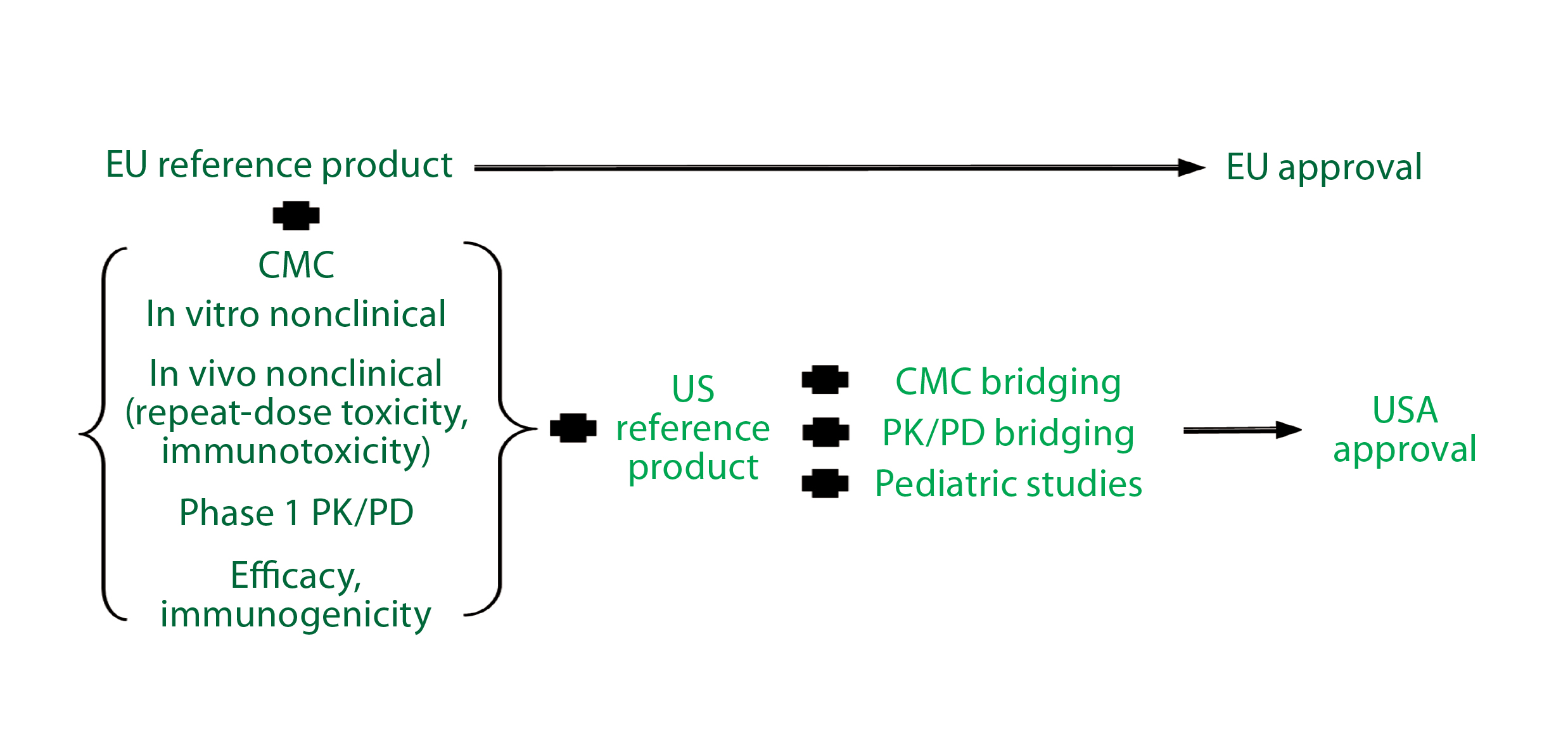
Figure 2: Reference standard requirements for biosimilar development in major markets
According to the EMA biosimilar guidelines (Figure 2), if an applicant performs parallel development for Europe and the United States, then inclusion of US reference standards is necessary. Scientifically, the type of bridging data needed always will include data from analytical studies (e.g., structural and functional data) that compare all three products (the proposed biosimilar, the EU reference product, and the US comparator). They may include data from clinical pharmacokinetic (PK) and/or pharmacodynamic (PD) bridging studies for all three products as well.
Extensive Comparability Data: Biological systems are inherently variable, and expression systems can substantially affect the structure and function of proteins they produce. Thus, biological products are extensively characterized for variability, even among different lots of the same product. Stringent EMA and FDA regulations require comprehensive structural and functional analytic comparative data to demonstrate comparability before initiating preclinical testing and clinical PK/PD studies (10).
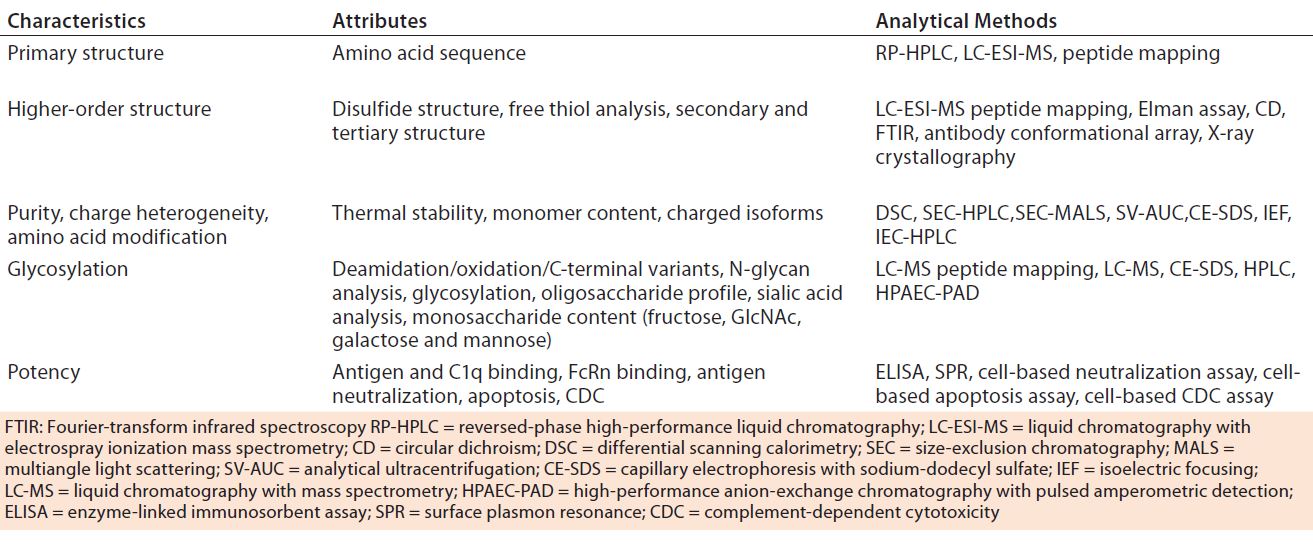
Table 5: Physicochemical and biological characterization methods for comparability studies of biosimilars (8)
Biomolecular analyses fall under three categories: physicochemical, immunological, and biological assays. Table 5 summarizes those used in comparability studies of biosimilar and reference products. Demonstrating a high level of analytical similarity between those drugs is the first step. All structural elements and modifications of a protein should be evaluated with full capability of detecting differences. Based on observed characteristics of the reference product, a quality target product profile (QTPP) is defined for a biosimilar.
Control Strategy: The term “control strategy” refers to a combination of input, procedural, and testing controls that ensure that a bioprocess consistently delivers products that meet quality attribute requirements. The level of control needed for each individual quality attribute is determined based on the criticality level of that attribute and the capability of a process to deliver product consistently that meets quality expectations.
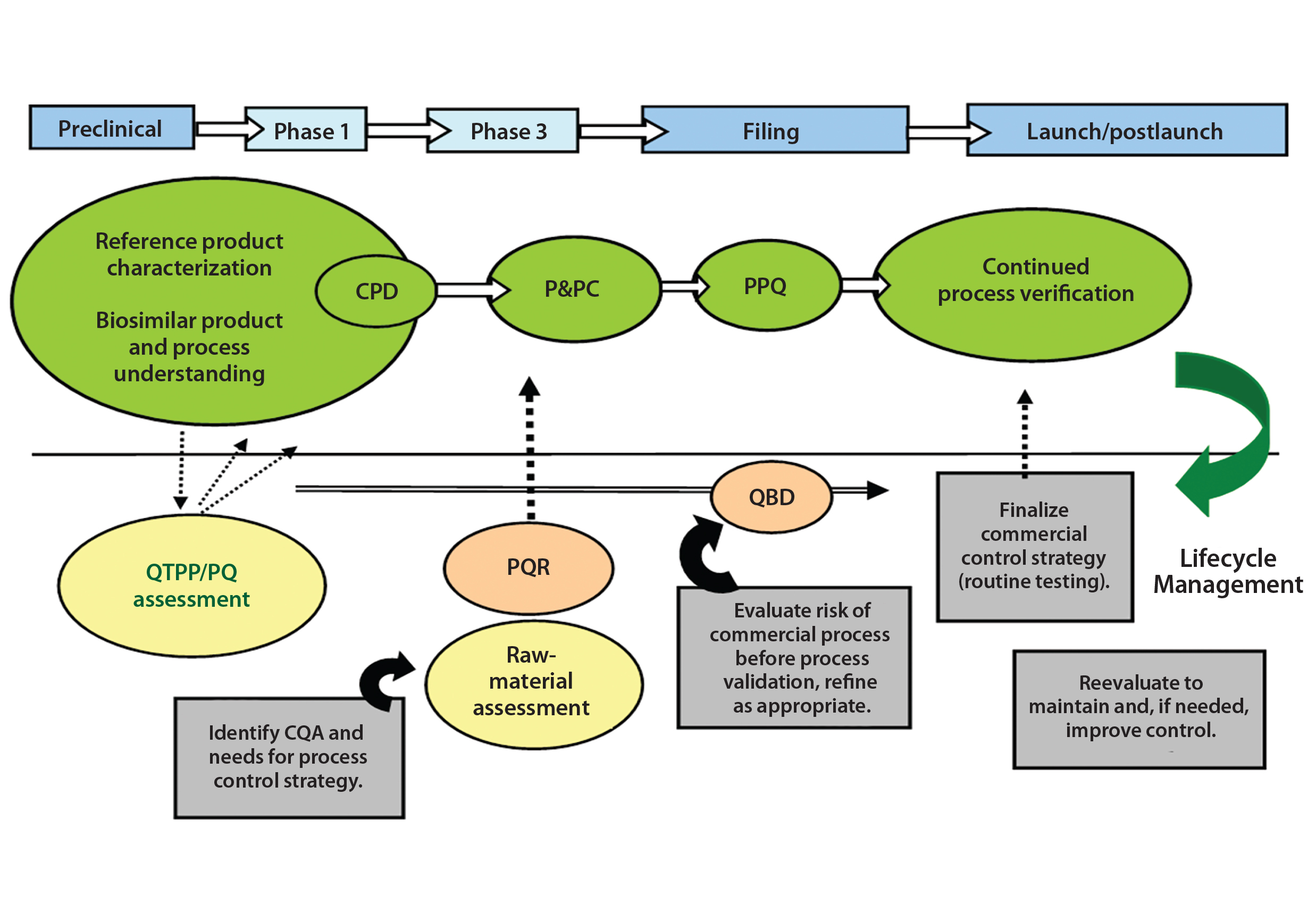
Figure 3: Control strategy during biosimilar development
An integrated control strategy includes procedural and raw-material controls, in-process control (IPC) tests, process monitoring and product data monitoring, release specification testing, stability testing, process validation, characterization testing, control of process validation, and comparability testing. Figure 3 illustrates development of a control strategy and its importance to product lifecycle management.
Establishment of specifications is a major area of uncertainty for a biosimilar integrated control strategy. Regulatory expectations for commercial specifications are not completely clear, however. EU guidance mentions that selection of tests to be included in specifications (or control strategy) is product specific for both drug substance and drug product, and thus should be defined as described in ICH Q6B (11). US guidance, however, offers no specific reference to expectations for development of a biosimilar control strategy.
Manufacturing Changes and Challenges: Some biosimilar manufacturers may need to make changes or alter their own manufacturing processes — for enhancement of product quality and yield, adherence to upgraded regulatory policies, or increased efficiency and improved reliability of the biomanufacturing process (12). Modifications to any biological product — whether originator or biosimilar — that result from manufacturing changes are subject to tight regulatory controls and limits (13). Table 6 illustrates such changes and associated regulatory requirements during different stages of manufacturing.

Table 6: Manufacturing changes and regulatory recommendations
Clinical Development: Clinical trials for biosimilars must demonstrate safety and efficacy comparable to their comparators through sequential PK/PD, immunogenicity, and efficacy/safety trials. Stand-alone phase 3 studies or combined phase 1–3 designs without supporting PK data are unlikely to be accepted by regulatory reviewers. Subject to a risk-based approach, clinical comparability requirements vary by case. Three-arm phase 1 clinical trials can demonstrate comparability for a biosimilar and two licensed versions of the same reference product on different markets (e.g., Europe and the United States). That allows developers to proceed with pivotal phase 3 trials using a single version of the reference product for biosimilar approval in each respective market (14). Extrapolation from one indication to another is allowed by both the EMA and FDA for biosimilars, although it must be supported by strong scientific justification (15).
Interchangeability
As with generic drugs, interchangeability would allow biosimilars to be substituted by pharmacists without the intervention of a prescribing doctor. This is the next big hurdle in the path of biosimilars, clearing which would create a real challenge to the reference products’ markets (16). Such substitution is what allows generic medicines to gain market share rapidly (17). However, some prescribers may have safety concerns.
Immunogenicity studies present an additional consideration, particularly for interchangeable biosimilars, although they generally are not required for manufacturing changes. But designation of interchangeability would allow pharmacists to substitute such a biosimilar for its reference like they do with small-molecule generic drugs (18). The candidate on which switching studies would be conducted must be the same as that approved without an interchangeability designation. There is no higher regulatory standard for biosimilars, and the data burden to obtain a designation of interchangeability is increased (7).
To be considered interchangeable in the United States, biosimilars must (among other things) “be expected to produce the same clinical result as the reference product in any given patient,” according to the Biologics Price Competition and Innovation Act of 2009. Developers also must document that a patient could switch back and forth between the original and biosimilar products without increased risk. According to the FDA’s rules, neither biosimilars nor interchangeable drugs can have clinically meaningful differences in safety when compared with the originator products (19).
Interchangeability and substitution of biosimilars are not within the scope of EU regulatory approval, so there is no agreed-upon definition of what interchangeability actually means to Europe and no inclusion of such information in the European Public Assessment Report (EPAR) (15). Currently, the United States is the only country allowing for a formal such designation among biologic products. A recent FDA draft guideline requires that sponsors show that a proposed product “is biosimilar to the reference product” (20). When a product is first licensed as a biosimilar, that licensure may be referenced to support the statutory criterion for demonstrating interchangeability on the basis of a switching study or studies.

Table 7: Recommendations for overcoming challenges to market access
Opportunities and Recommendations
Based on a literature review, we offer in Table 7 some recommendations to overcome barriers to the market access for biosimilar MAbs (21). Once such blockbusters start to go off patent in Europe, biosimilars should become 20–30% cheaper than original MAb products. Market competition will play a pivotal role in favor of biosimilars. These speculations come from the fact that a 30–40% price reduction already has been seen for epoetin and filgrastim (granulocyte colony-stimulating factor). The sole purpose of biosimilar development is to lower the cost relative to original biologics and take their market share.
IMS Health forecasts that the global biologics market should reach $250 billion by 2020. Until then, some blockbuster MAbs will continue to dominate because of their patent protection. Once biosimilar MAbs start to invade their market, however, the scenario will change dramatically. Biosimilars and nonoriginal biologics are likely to take 4–10% market share by 2020, worth about $10–25 billion. That anticipation hinges on the number of biosimilars, especially those introduced to the US market. The United States represents the largest potential market for biologics because of its high per capita consumption of biologics overall.
By 2020, the 12 products that share 40% of the current global biologic market (worth $72 billion in sales) will face patent expiration. Such a goldmine will be too lucrative for biosimilars to be kept away, and the race has begun already. In September 2013, the EMA approved the first two biosimilar versions of Remicade (infliximab). Now, those two biosimilars together incur more sales than all the other biosimilars on the market combined (12). With the great majority of products in development targeted for US, EU, and other major markets, the current pipeline includes ~800 biosimilars and ~500 biobetters (a total of ~1,300 follow-on products) in development pipeline for >100 currently marketed biopharmaceutical reference products (22). The IMS Health report provides an extensive study of the rapidly growing market. As the biotechnology industry strives to minimize costs, biosimilars are viewed as viable substitutes to highly expensive innovator biological medications (23).
Demand for biologics is increasing in many healthcare sectors, more so than for small molecules. But the biosimilar landscape is replete with landmines of complications associated with regulatory, manufacturing, bioanalytical, and marketing concerns. Outsourcing could lead to cost-effective development while minimizing some challenges. And all can be overcome with an in-depth knowledge of each region, early strategic planning, and effective communication with regulatory agencies. Successful development and commercialization of biosimilars requires business strategies that integrate appropriate clinical design and regulatory compliance.
Although it requires a substantial investment in time and money, the development and introduction of biosimilars ultimately should provide cost savings compared with innovator products. The growth rate for biosimilar MAbs might be over 25% by 2020. The industry of biosimilars should bring benefits both for science and for healthcare.
References
1 Ginestro M, Moore J. How to Compete and Win in a World with Biosimilars: Commercial Launch Strategies and Defensive Positioning for the US. KPMG: Chadds Ford, PA, 2015.
2 Acha V. Countering Points Made in Biosimilars. Pharmaceut. J. 30 Jan 2017; www.pharmaceutical-journal.com/opinion/correspondence/countering-points-made-in-biosimilars-piece/20202218.article.
3 Rader RA. Manufacturing Costs Will Be Critical to Biosimilars’ Success. Pharmaceut. Manufact. 8 November 2016.
4 Rémuzat C. Key Drivers for Market Penetration of Biosimilars in Europe. J. Market Access Health Pol. 5(1) 2017; doi:10.1080/20016689.2016.1272308.
5 Biosimilars Approved in Europe. GaBI J. 24 February 2017; www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe.
6 Jensen AR. US FDA Proposals for Naming of Biologicals and Labelling of Biosimilars. GaBI J. 5(3) 2016: 140–143.
7 CBER/CDER. Nonproprietary Naming of Biological Products: Guidance for Industry. US Food and Drug Administration: Rockville, MD, January 2017.
8 Tsuruta LR, Santos ML. Biosimilar Advancements: Moving on to the Future. Biotechnol. Progr. 31(5) 2015: 1139–1149; doi:10.1002/btpr.2066.
9 CHMP/437/04 Rev 1. Guideline on Similar Biological Medicinal Products. European Medicines Agency: London, UK, 23 October 2014.
10 Navaneethaselvan D, et al. The Opportunities and Challenges Involved in Registration of Similar Biotherapeutic Products in Emerging Countries. Int. J. Res. Pharmacy Sci. 3(3) 2013: 6.
11 ICH Q6B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. US Fed. Reg. 18 August 1999: 44928; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf.
12 Islam MS, Alam MU, Amin M. A Global Perspective of the Prospects and Challenges of an Awaiting Revolution of Biosimilars. Biojournal Sci. Technol. 2, 2015: 7–16.
13 Biosimilarity and Comparibility After Manufacturing Changes: Can a Biologic Become a Biosimilar of Itself? European Biopharmaceutical Enterprises: Brussels, Belgium, 22 February 2016.
14 Hurley P, Borley J, Congiatu C. Challenges in Global Biosimilar Development: A Regulatory Perspective. Contract Pharma June 2015.
15 Macdonald JC, et al. Regulatory Considerations in Oncologic Biosimilar Drug Development. mAbs 7(4) 2015: 653–661; doi:10.1080/19420862.2015.1040973.
16 First Biosimilar with Extrapolation: Interchangeability Is Next. FDAMap 18 February 2016; www.fdamap.com/first-biosimilar-with-extrapolation-interchangeability-is-next.html.
17 Schueller T. Chances and Hurdles for Biosimilars. CHEManager Int. 8 October 2015; www.chemanager-online.com/en/topics/pharma-biotech-processing/chances-and-hurdles-biosimilars.
18 McCamish M, et al. Toward Interchangeable Biologics. Clin. Pharmacol. Ther. 97(3) 2015: 215–217; doi:10.1002/cpt.39.
19 Friedman LF. An Innovation That Could Transform the Drug Industry Faces a Major Hurdle. Business Insider 30 April 2015; www.businessinsider.com/biosimilars-bioequivalence-and-interchangeability-2015-4.
20 Draft Guidance for Industry: Considerations in Demonstrating Interchangeability with a Reference Product. US Food and Drug Administration: Rockville, MD, January 2017; www.gpo.gov/fdsys/pkg/FR-2017-01-18/pdf/2017-01042.pdf.
21 Moorkens E, et al. Overcoming Barriers to the Market Access of Biosimilars in the European Union: The Case Study of Biosimilar Monoclonal Antibodies. Front. Pharmacol. 7, 2016: 193; doi:10.3389/fphar.2016.00193.
22 Rader RA. Biosimilars Pipeline Analysis: Many Products, More Competition Coming. BioProcess Online 26 July 2016.
23 Biosimilar Market Is Projected to be Worth USD 32 Billion Worldwide by 2025 According to “Global Biosimilars Market, 2015–2025.” GlobeNewswire: El Segundo, CA, 12 November 2015.
24 A Health-System Pharmacist’s Guide to Biosimilars: Regulatory, Scientific, and Practical Considerations. American Society of Health-System Pharmacists: Bethesda, MD, 2013; http://ashpadvantagemedia.com/downloads/biosimcentral_guidelines.pdf.
25 ICH Q11: Development and Manufacture of Drug Substances. Federal Register 20 November 2012, Vol. 77, No. 224, p. 69634-5; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf.
Pankaj S. Chaudhari is regulatory affairs and quality assurance (RA/QA) manager (pankaj.chaudhari@ipca.com), Rajalaxmi Nath is RA/QA research associate, and corresponding author Sanjeev K. Gupta is general manager of the advanced biotech laboratory at Ipca Laboratories, Ltd. in Mumbai, India; +91-22-6210-5820; sanjeev.gupta@ipca.com; www.ipca.com.
I want doi or pcmid for this
dede bhai