
Figure 1: Refolding analysis of serum-derived human IgG after 8 M urea treatment; (A) spike study with IgG derived from human serum — urea-treated IgG spiked into native IgG solution at 0.1% and 1% and compared with native IgG only sample; (B) spike study with IgG derived from human serum — urea-treated IgG spiked into native IgG solution at 10% and 100% and compared with native IgG only sample. Note: Not until 100% unfolded antibody is added do we observe HOS differences, which suggests that IgGs derived from human serum have a much faster refolding capability than CHO-derived proteins. Error bars show assay variation.
For protein therapeutics and other biologics, the importance of the molecule’s structure to its efficacy and safety is well established (1–5). In particular, their tertiary and quaternary structures play very important roles in product quality and have been monitored extensively in comparability studies (6–12). However, because of both the large molecular size and rotational property of amino acid α carbons, a protein can assume an enormous number of different conformations (13). For antibody-based biologics such as monoclonal antibodies (MAbs), fusion proteins, and antibody–drug conjugates (ADCs), an additional degree of conformational change can be created by the relatively free Fab and Fc domains.
The complexity of antibody higher order structure (HOS) makes accurate determination of its status a challenge. A number of technologies have been used to analyze MAb HOS: e.g., circular dichroism (CD), Fourier-transform infrared (FTIR) analysis,
fluorescence spectroscopies, and differential scanning calorimetry (DSC) (8, 12, 14, 15). Even though their principles of measurement have been well established scientifically for some aspects of protein HOS, they all have limitations. For example, read-outs are rarely associated with accurate and individual structural assignment; instead, they provide an average of the whole population analyzed. Recent studies also indicate that some of these technologies cannot detect HOS differences for MAbs under selected stress conditions (1, 11, 16–18).
Monoclonal antibodies (MAbs) are the largest and most rapidly growing class of biologics. More than 60 MAbs have been approved so far, including many of the best-selling drugs in the world (19). Even though great success has been achieved in this product class overall, many MAbs still fail in clinical development — some for safety reasons such as immunogenicity (1, 5, 20–22). It is therefore important to develop new technologies that can sensitively and accurately measure MAb HOS and provide useful information for making the right bioprocess and formulation choices during the MAb development and production.
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) is a new technology for monitoring protein HOS based on time-dependent hydrogen–deuterium exchange of amide groups on the molecular surface, the rate of which depends on both the degree of exposure and a protein’s immediate environment (23, 24). By measuring that exchange rate, analysts can compare the status of a protein’s HOS. This technology also has its limitations: For example, a protein needs to be quenched at lower pH, followed by digestion and chromatographic separation before mass spectrometry can be applied to identify the resulting fragments. That complex process makes the technology useful primarily for protein characterization and small-scale comparability studies.
For high-volume analytical assays such as those needed in cell-line selection and development of bioprocesses and formulations, more accurate and relatively high-throughput methods are desirable. Protein conformational array (PCA) technology was developed based on the epitope-scanning principle in immunology (25). Using a large array of antibodies that covers a whole MAb molecule and measuring MAb surface epitope exposure, HOS can be described precisely and systematically. Thus, PCA is in an enzyme-linked immunosorbent assay (ELISA) format that is easy to use with high precision and accuracy (26). It has been used successfully in biosimilar and novel MAb bioprocess development (27–29). In MAb development, one goal is to produce an antibody that resembles antibodies from human blood so that potential drug immunogenicity can be minimized. From the early generation of murine MAbs with high immunogenicity to the chimeric, humanized, and finally totally human MAbs, that immunogenicity potential has been greatly reduced over the years. However, clinical reports show that many such products still present immunogenicity concerns (4, 5, 17, 28, 29).
One interesting question is what role cell line, bioprocess, and formulation decisions play in producing a MAb that is highly similar to those from human serum. We compared IgGs purified from human serum with three different biologics containing CH2 and CH3 domains, testing their HOS stability under four different stress conditions. Our results indicated that biologics derived from Chinese hamster ovary (CHO) cells display HOS stability polymorphism. Each molecule responded to stress conditions very differently even in its same CH2–CH3 domains. The significance of that finding to biologics development is discussed below.
Materials and Methods
Reagents: All chemicals came from MilliporeSigma. We bought 96-well microplates from Corning and both Streptavidin–HRP conjugate and biotin labeling kits from Thermo Fisher Scientific.
Antibodies and ELISA Kits: All antibodies and ELISA kits used in this study are products of Array Bridge. Polyclonal antibodies against the peptides were produced by New Zealand white rabbits. For the sandwich ELISA, we coated antibodies against each region of the MAb molecule on 96-well plates in different columns; with each antibody coating six wells in rows B–G of the same column. We used a biotin-labeled rabbit antihuman IgG antibody as the reporting antibody to detect MAb–peptide antibody complexes and streptavidin–HRP to detect complexes formed by antihuman IgGs, MAbs, and antipeptide antibodies. Signal strength of the sandwich ELISA depends on relative epitope exposure of a MAb in each region. If more epitopes from the MAb can be recognized by the peptide-derived antibody, then a stronger signal is produced — and vice versa.
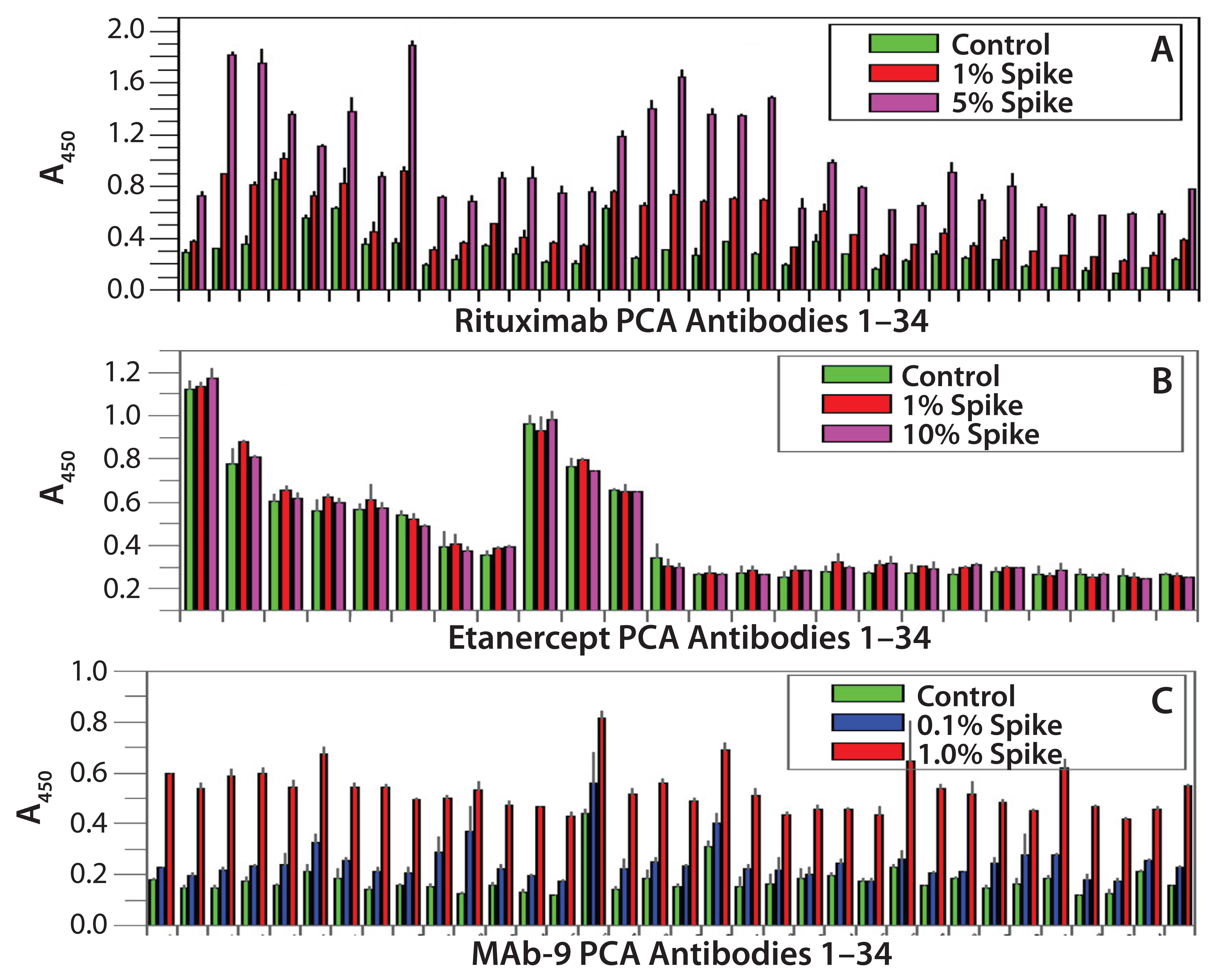
Figure 2: Refolding analysis of rituximab, etanercept, and MAb9 after 8 M urea treatment
Sample Treatment: For ureadenatured MAbs, we added urea at a final concentration of 8 M to a 10-mg/mL MAb solution and incubated that overnight at 4 °C. Subsequently we spiked the unfolded MAb solution into a native-MAb solution at the percentages indicated in Figures 1 and 2, with a final protein concentration of 5 µg/mL for ELISA analysis. All samples — rituximab, etanercept, human IgG, and MAb 9 — were treated at similar protein concentrations for 16 hours under different conditions before being stored at 4 °C until testing:
- high temperature (55 °C)
- oxidation (adding 1/10 volume of 30% H2O2)
- low pH (adding 1/10 volume of 1M HCl for a final pH of 3.0)
- high pH (adding 1 volume of 1M Tris for a final pH of 9.5)
- control (no treatment, stored at 4 °C).
For reporting antibodies, we developed a polyclonal antihuman IgG antibody to detect human-derived IgGs captured by the coating antibody. We labeled our reporting antibody with biotin, which in a subsequent step forms a complex with streptavidin–HRP conjugate. We used TMB (3,3′,5,5′-tetramethylbenzidine) as a substrate for the HRP enzyme-activity assay. Following a short time to allow color formation from the enzymatic activity, we added an equal volume of 1M sulfuric acid to stop the reaction and used a VersaMax spectrophotometer from Molecular Devices to measure the color change at 450 nm.
Results
Refolding Dynamics of Human-Serum– Derived IgGs: For this experiment, we used IgG molecules purified from human serum to test their refolding dynamics in our PCA ELISA. To unfold the IgGs overnight, we used 8 M urea. Then we spiked the unfolded IgG into native IgG preparations from 0.1% to 100% (the latter meaning that half of the testing IgGs at 5 µg/mL were from the urea-treated sample and the other half was from native IgGs). Figures 1A and 1B indicate that IgGs derived from human serum refolds much more quickly than other IgGs tested (Figure 2). Those results indicate that the PCA ELISA did not detect obvious new-epitope exposure with ≤30% spike of unfolded IgGs, whereas as low as a 0.1% spike of unfolded molecules could be detected from CHO-derived MAbs (25).
Refolding Dynamics of CHO Cell-Derived MAbs and Fusion Proteins: We selected three CHO cell-derived proteins to test their folding properties with the PCA ELISA: rituximab (IgG1), etanercept (a fusion protein of TNF-α receptor with human IgG1 CH2-CH3 domains), and an IgG2 MAb that is currently under clinical development (MAb 9). Figure 2 shows that etanercept has the fastest refolding process very similar to that of the serum-derived IgGs (Figure 1), whereas MAb 9 showed the slowest refolding. A 0.1% spike (5 ng/mL of urea-treated MAb into 5 µg/mL native MAb) can be quantified based on the new epitope exposure. Rituximab has an intermediate rate of refolding, with a 1% spike quantified by the PCA ELISA.
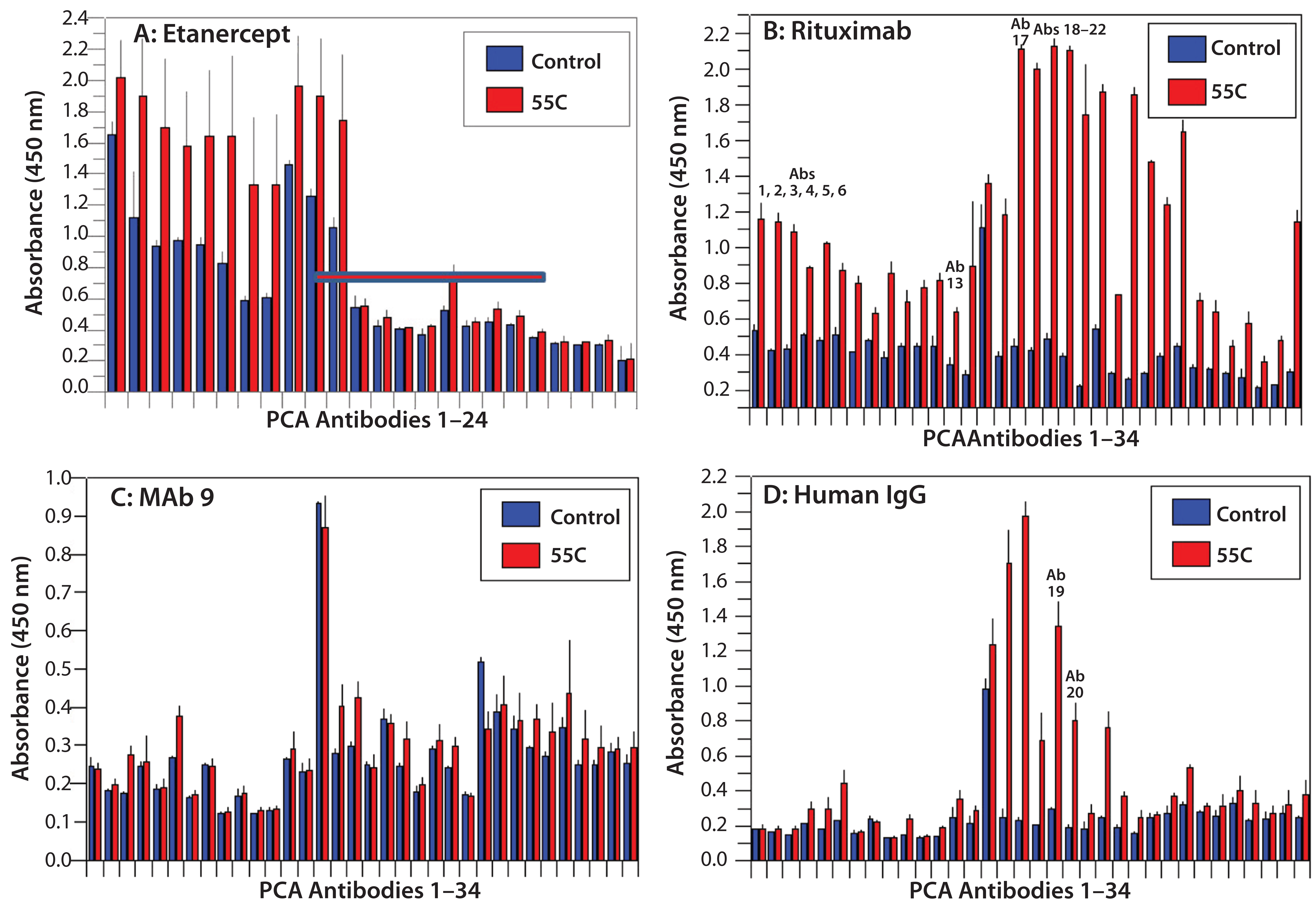
Figure 3: HOS stability of four CH2- and CH3-containing proteins under temperature stress
HOS Stability at Elevated Temperature: In biologics formulation development, elevated temperature is used in accelerated stability testing to predict the products’ real-world stability (11, 30–32). We compared the above-listed molecules of different modality or derived from different sources for their HOS stability. Rituximab proved to be most sensitive to elevated temperature: Increased epitope exposure was detected with all 34 capturing antibodies produced from 34 overlapping peptides covering the whole MAb molecule (Figure 3B). However, MAb 9 demonstrated the most HOS stability at elevated temperature: Only minor increases (new epitope exposure) or decreases (inward movement of epitopes) of change were detected from different regions of the molecule (Figure 3C). Etanercept showed some instability in its TNF-α receptor domain, whereas IgGs derived from human serum showed some instability in their hinge regions and to a lesser extent in their light-chain constant region and heavy-chain CH1 domain (Figures 3A and 3D).

Figure 4: HOS stability of four CH2- and CH3-containing proteins after oxidation
HOS Stability After Protein Oxidation: Protein oxidation can affect the HOS status of MAb molecules (33, 34). We introduced protein oxidation by hydrogen-peroxide treatment of our four selected proteins. The results indicated that etanercept was the most stable molecule to oxidation, whereas MAb9 was the least stable in its HOS after protein oxidation (Figures 4A and 4C). This presents an interesting contrast with what we found at elevated temperatures, in which MAb 9 was the most stable molecule (Figure 3C). IgGs derived from human serum demonstrated relatively good HOS stability during oxidation, their only change being observed in the CH1 domain of the heavy chain (Figure 4D, Ab19 and Ab20). For rituximab, oxidation affected both the variable and constant regions, especially in the lightchain Fv domain (Figure 4B, Ab1 to Ab6) and constant domain (Ab13 to Ab17) and the heavy-chain CH1 domain (Ab18 to Ab22).

Figure 5: HOS stability of four CH2- and CH3-containing proteins at acidic pH
HOS Stability Under Acidic pH: During MAb purification, a pH <4 is used in the protein A affinity column step to elute purified MAbs, which then are held at the low pH condition for some time to inactivate potential viruses. So it is important to test MAb HOS stability under acidic conditions. We incubated our four proteins at pH 3 overnight and determined their HOS status using PCA ELISA.
Under these conditions, MAb 9 proved to be the least stable protein, with dramatic increases in new-epitope exposure across the entire molecule indicating a combination of systematic unfolding and regional changes (Figure 5C). Etanercept and rituximab, however, demonstrated more HOS stability under acidic conditions — especially etanercept, for which no HOS changes were detected, whereas rituximab showed some regional changes in epitope exposure (Figures 5A and 5B). IgGs derived from human serum also showed HOS instability, especially in their constant regions (Figure 5D).
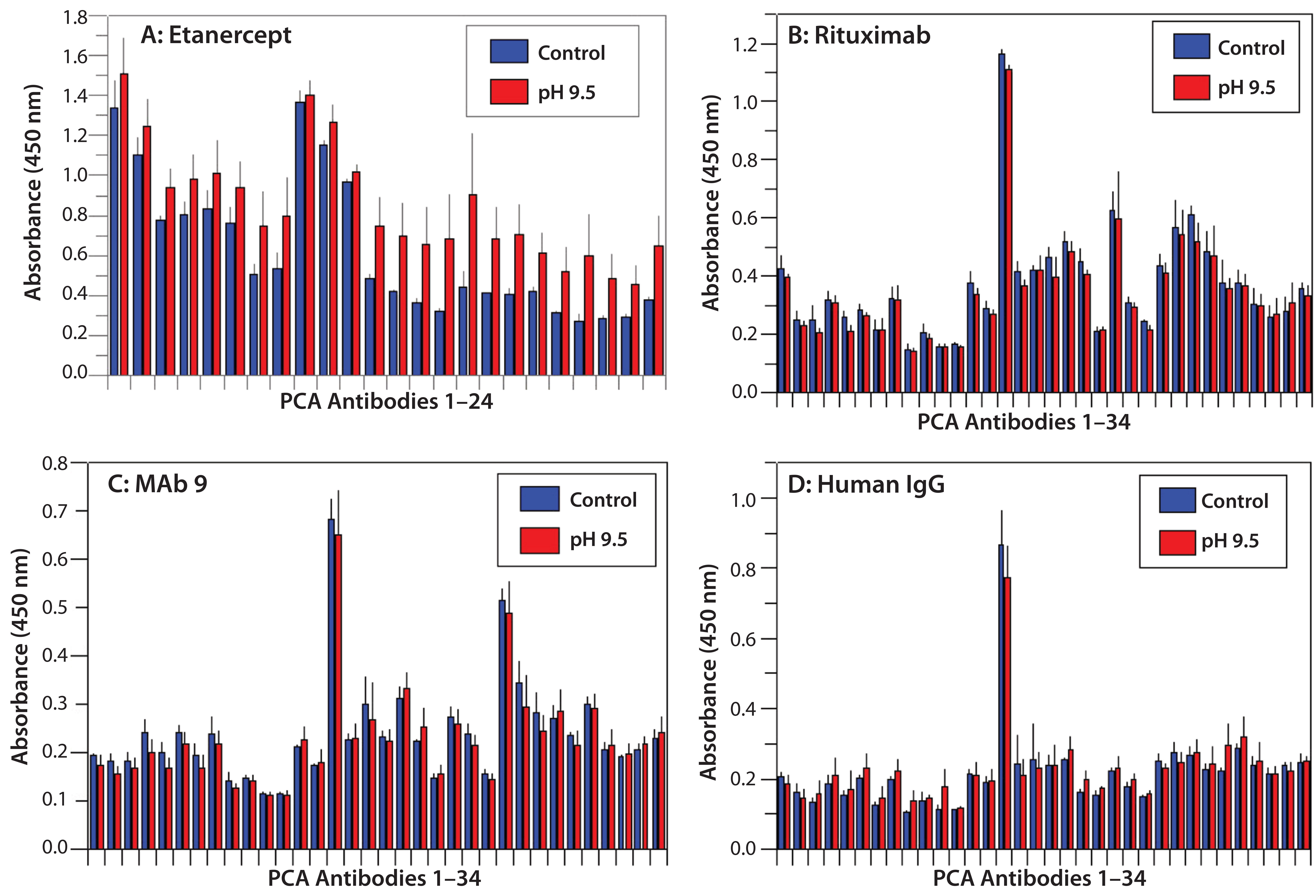
Figure 6: HOS stability of four CH2- and CH3-containing proteins at basic pH; note that higher pH does not affect HOS.
HOS Stability at Basic pH: Previous studies have shown that proteins undergo deamidation under basic pH conditions (35). We incubated our four different proteins at pH 9.5 and tested their HOS stability with PCA ELISA (Figure 6). All four showed good HOS stability at high pH, which is in clear contrast with results from the acidic treatment above.
Discussion
Anfinsen’s classic study of ribonuclease A indicated that the native structure of small globular proteins is determined only by their amino acid sequence (36). Later studies of molecular chaperones demonstrated that many proteins (e.g., Hsp60, Hsp70, and Hsp90) require their assistance in vivo (37). It is believed that chaperones function to assist protein refolding, but the final state of such proteins does not appear to be affected by the chaperones — except for prion proteins, which have stable conformations that are different from the native folding state.
A previous PCA study of MAb HOS have showed that each MAb has a unique and different HOS signature, which is stable from batch to batch unless a process deviation occurs (25). Another study demonstrated that HOS changes induced by both physical and chemical stress conditions (e.g., temperature, oxidation, glycosylation, and pH extremes) all could be detected and quantified using PCA technology (26). Amino acid sequence alignment analysis indicated that for all human MAbs on the market and in the protein sequence database, both the CH2 and CH3 domains are highly conserved (38).
One goal in MAb development is to mimic human IgGs from serum to achieve minimum immunogenicity and maximum efficacy and stability. CHO cell lines have been used to produce most MAbs on the market and in clinical development; however, many marketed MAbs still showed different degrees of immunogenicity (17, 36, 37, 39, 40). It is therefore interesting to analyze the HOS stability of CHO-derived proteins with the same CH2 and CH3 domains and compare the results with those of IgGs purified from human serum.
Here, we compared the refolding process of human-serum–derived IgG and three other CHO-derived proteins with CH2 and CH3 domains. Note that after 8 M urea treatment, both the human serum-derived IgGs and etanercept showed very fast refolding (within a few minutes) as monitored by the PCA ELISA. By contrast, one IgG1 molecule (rituximab) and an IgG2 molecule (MAb 9) showed slow refolding, reflected by the detection of significant new epitope exposure after 8 M urea treatment (Figures 1 and 2).
For human-serum–derived IgGs, only a 100% spike showed a small increase in new epitope exposure (Figure 1B). MAb 9 showed the slowest refolding rate. As low as 0.1% spike of the urea-treated IgGs will show a detectable increase in new epitope exposure (Figure 2C). Rituximab showed a similar increase with a 1% spike (Figure 2A). These results demonstrate that CHO-derived proteins can have very different HOS stability properties from those of human-serum IgGs. They also show that CHO cells can produce fast-refolding proteins with stable HOS, as in the case of etanercept.
Because the CH2 and CH3 domain amino acid sequences of all four human IgG molecules are highly conserved, it is not clear whether the TNF-α receptor on the etanercept is involved in stabilization of the protein HOS or different CHO cell lines used for its production play a role. Another possibility is that amino acid differences between human-serum–derived IgG and rituximab/MAb 9 affect their refolding property. Sequence comparison of a large number of fast- and slow-refolding antibodies should help to answer this question.
A more interesting question is whether a MAb’s refolding dynamics serve as an important indicator of its immunogenicity potential. Rituximab has had immunogenicity issues in clinical use, and more epitope exposure was detected from rituximab than with other Mabs on the market. TNF-α and IL-6 were induced after rituximab treatment, whereas no serious immunogenicity issues have been reported with etanercept (41). More studies on refolding dynamics of marketed MAbs should provide more information.
In a further test of HOS stability, we applied different stress conditions to our four proteins. Under elevated temperature, MAb 9 showed the most HOS stability as analyzed by the PCA ELISA, whereas rituximab had the least stability. For the former, only three regions showed minor epitope exposure at elevated temperature (Figure 3C), whereas the latter exhibited new epitope exposure across the whole antibody array, with the most changes in its CH2 domain (Figure 3B). That finding is in agreement with previous studies using DSC, which showed CH2 to be the first domain to undergo unfolding, whereas CH3 is unfolded in the second transition at a higher temperature (12). IgGs from human serum also showed unfolding at 55 °C, even though their unfolding mainly occurred in the CH1 and CH2 domains of their heavy chains (Figure 3D). For etanercept and MAb 9, the CH2 and CH3 domains were stable at 55 °C, reflecting a higher melting temperature, whereas the TNF-α receptor domain in etanercept underwent some unfolding (Figure 3A).
Efficacy and stability of protein molecules can be compromised by oxidation (13). DSC can detect the resulting HOS changes, whereas other orthogonal methods (e.g., near-UV CD and fluorescence spectroscopy) are far less sensitive in doing so (18). Here, we demonstrated that a PCA ELISA can detect oxidation-induced HOS changes from all four tested proteins (Figure 4).
Note that PCA ELISA technology also quantifies HOS changes while also pointing to affected molecular regions. For example, etanercept exhibited only minor HOS changes, mainly in the TNF-α receptor domain, whereas IgGs from human serum showed changes at the CH2 domain (where methionine oxidation primarily occurs). Rituximab showed HOS changes in both its variable region and its CH1 and CH2 domains, whereas the CH3 domain was less affected (Figure 4B). HOS changes occurred across the whole MAb-9 molecule, suggesting that protein oxidation has a major effect on it (Figure 4C).
Because low-pH solutions are part of protein A affinity-column purification and virus inactivation, we also tested the four proteins’ HOS stability under acidic conditions. Figure 5 shows that etanercept and rituximab were relatively stable under acidic conditions. with only a few regions from rituximab affected (coating antibodies Ab1, Ab19, and Ab34 corresponding to the light-chain Fv, CH1, and CH3 domains, respectively). On the other hand, MAb 9 and serum-derived IgGs were highly sensitive to acidic pH treatment. Both showed major changes in their HOS status, especially MAb9 (which showed signs of systematic unfolding instead of just regional changes).
Finally, we incubated the four proteins at basic pH conditions, with which all four showed relatively stable HOS. Only etanercept showed minor HOS changes with high pH. This might reflect the possibility that protein deamidation is a slow process. Some literature examples tested high-pH solutions for several days or longer (21), whereas in our study, all such treatments lasted overnight only.
A Possible Predictor of Performance
We used PCA ELISA to test HOS stability of four proteins (three recombinants, and one derived from human serum). Our results indicate a diverse response to different stress conditions among the four proteins tested. IgGs from human serum and etanercept have the fastest refolding capability, whereas MAb 9 showed the slowest refolding after urea treatment.
The fact that all four proteins contain human-IgG CH2 and CH3 domains but responded to unfolding and stress very differently raised an interesting question: What factor(s) determine CH2 and CH3 HOS stability? Is it the very few amino acid residuals that are different among the four proteins, or is it the host cells (CHO or human B cells) from which they are expressed? How much influence do bioprocess details have on stability of CHO-derived proteins? Could unfolding–refolding testing of the urea-induced HOS changes be a predictor for potential immunogenicity? Rituximab has induced cytokine release in clinical use and in whole-blood assays (41). Is it possible to establish a correlation between a MAb’s HOS stability and its immunogenicity potential?
Because the IgGs derived from human serum are a mixture of the four polyclonal subclasses, it is unclear from our study which subclass(es) responded to the different stress conditions. But it is obvious that all IgG subclasses refold very fast after urea denaturation. With recent developments in immunotherapy, more IgG4 molecules are in clinical development. It will be interesting to test those from human serum and compare them with CHO-derived corresponding IgGs for HOS stability. The goal would be to evaluate whether PCA ELISA technology can be used in monitoring the HOS status of other classes of IgG candidates and whether it can predict their clinical immunogenicity based on the HOS stability property.
References
1 Bessa J, et al. Immunogenicity of Antibody Aggregates in a Novel Transgenic Mouse Model. Pharma. Res. 32(7) 2015: 2344–2359; doi:10.1007/s11095-015-1627-0.
2 Buttel I, et al. Taking Immunogenicity Assessment of Therapeutic Proteins to the Next Level. Biologicals 39(2) 2011: 100–109; doi:10.1016/j.biologicals.2011.01.006.
3 Hermeling S, et al. Structural Characterization and Immunogenicity in Wild-Type and Immune Tolerant Mice of Degraded Recombinant Human Interferon Alpha2b. Pharma. Res. 22(12) 2005: 1997–2006.
4 Hermeling S, et al. Structure-Immunogenicity Relationships of Therapeutic Proteins. Pharma. Res. 21(6) 2004: 897–903.
5 Jiskoot W, et al. Immunological Risk of Injectable Drug Delivery System. Pharma. Res. 26(6) 2009: 1303-1314; doi:10.1007/s11095-009-9855-9.
6 Dyson H, Wright P. Peptide Conformation and Protein Folding. Curr. Opin. Struct. Biol. 3, 1993: 60–65.
7 Dyson H, Wright P. Defining Solution Conformation of Small Linear Peptides. Annu. Rev. Biophys. Chem. 20, 1991: 519–538.
8 Gabrielson JP, Weiss WF. Technical Decision-Making with Higher Order Structure Data: Starting a New Dialogue. J. Pharm Sci. 104(4) 2015: 1240-1245; doi:10.1002/jps.24393.
9 Harris RJ, Shire SJ, Winter C. Commercial Manufacturing Scale Formulation and Analytical Characterization of Therapeutic Recombinant Antibodies. Drug Dev. Res. 61(3) 2004: 137–154; doi:10.1002/ddr.10344.
10 ICH Q5E. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. US Fed. Reg. 70(125) 2005: 37861–37862.
11 Zurdo J. Developability Assessment As an Early De-Risking Tool for Biopharmaceutical Development. Pharm. Bioprocess. 1(1) 2013: 29–50.
12 Jiang Y, et al. Technical Decision Making with Higher Order Structure Data: Higher Order Structure Characterization During Protein Therapeutic Candidate Screening. J. Pharm. Sci. 104(4) 2015: 1533–1538; doi:10.1002/jps.24406.
13 CBER/CDER. Guidance for Industry on Quality Considerations in Demonstrating Biosimilarity to a Reference Protein Product. US Food and Drug Administration: Rockville, MD, 2015.
14 Arthur KK, Dinh N, Gabrielson JP. Technical Decision Making with Higher Order Structure Data: Utilization of Differential Scanning Calorimetry to Elucidate Critical Protein Structural Changes Resulting from Oxidation. J. Pharm Sci. 104(4) 2015: 1548–1554; doi:10.1002/jps.24313.
15 Weiss WF, et al. Technical Decision Making with Higher Order Structure Data: Perspectives on Higher Order Structure Characterization from the Biopharmaceutical Industry. J. Pharm. Sci. 105(12) 2016: 3465–3470. doi: 10.1016/j.xphs.2016.09.003
16 Poppe L, et al. Profiling Formulated Monoclonal Antibodies By (1)H NMR Spectroscopy. Anal. Chem. 85(20) 2013: 9623–9629; doi:10.1021/ac401867f.
17 Ratanji KD, et al. Immunogenicity of Therapeutic Proteins: Influence of Aggregation. J. Immunotox. 11(2) 2014: 99–109; doi:10.3109/15 47691X.2013.821564.
18 Thiagarajan G, et al. A Comparison of Biophysical Characterization Techniques in Predicting Monoclonal Antibody Stability. MAbs 8(6) 2016: 1088–1097; doi:10.1080/19420862.201 6.1189048.
19 Walsh G. Biopharmaceutical Benchmarks 2014. Nature Biotech. 32(10) 2014: 992–1000; doi:10.1038/nbt.3040.
20 Hermeling S, et al. Antibody Response to Aggregated Human Interferon Alpha2b in Wild-Type and Transgenic Immune Tolerant Mice Depends on Type and Level of Aggregation. J. Pharm Sci. 95(5) 2006: 1084–1096; doi:10.1002/jps.20599.
21 Maas C, et al. A Role for Protein Misfolding in Immunogenicity of Biophmaceuticals. J. Biolog. Chem. 282(4) 2007: 2229–2236; doi:10.1074/jbc.M605984200.
22 Sharma B. Immunogenicity of Therapeutic Proteins, Part 1: Impact of Product Handling. Biotech Adv. 25(3) 2007: 310–317. doi:10.1016/j.biotechadv.2007.01.005.
23 Beck A, et al. Characterization of Therapeutic Antibodies and Related Products. Anal. Chem. 85(2) 2013: 715–736; doi:10.1021/ac3032355.
24 Berkowitz SA, et al. Analytical Tools for Characterizing Biopharmaceuticals and the Implications for Biosimilars. Nat. Rev. Drug Discov. 11(7) 2012: 527–540; doi:10.1038/nrd3746.
25 Wang X, Li Q, Davies M. Development of Antibody Arrays for Monoclonal Antibody Higher Order Structure Analysis. Front. Pharmacol. 4, 2013: 103; doi:10.3389/fphar.2013.00103.
26 Davies M, et al. MAb Higher Order Structure Analysis with Protein Conformational Array ELISA. Brit. J. Pharma. Res. 7(6) 2015: 401–412; doi:10.9734/BJPR/2015/18952.
27 Davies M, et al. Biosimilar MAb In-Process Sample Higher Order Structure Analysis with Protein Conformational Array ELISA. Brit. J. Pharma. Res. 9(3) 2016: 1–11.
28 Viitanen PV, et al. Mammalian Mitochondrial Chaperonin 60 Functions As a Single Toroidal Ring. J. Biol. Chem. 267(2) 1992: 695–698.
29 Wang X, Tabita FR. Interaction of Inactivated and Active Ribulose 1,5-Bisphosphate Carboxylase/Oxygenase of Rhodobacter sphaeroides with Nucleotides and the Chaperonin 60 (GroEL) Protein. J. Bacterial. 174(11) 1992: 3607–3611.
30 Arosio P, et al. Aggregation Stability of a Monoclonal Antibody During Downstream Processing. Pharm. Res. 28(8) 2011: 1884–1894. doi:org/10.1007/s11095-011-0416-7.
31 Correia I. Stability of IgG Isotypes in Serum. MAbs 2(3) 2010: 221–232.
32 Vermeer A, Norde W. The Thermo Stability of Immunoglobulin: Unfolding and Aggregation of a Multi-Domain Protein. Biophys. J. 78(1) 2000: 394–404; doi:10.1016/S0006-3495(00)76602-1.
33 Anfinsen CB, Haber E. Studies on the Reduction and Re-Formation of Protein Disulfide Bonds. J. Biol. Chem. 236, 1961: 1361–1363.
34 Schellekens H. Factors Influencing the Immunogenicity of Therapeutic Proteins. Nephrol. Dial. Translant. 20(S6) 2005: vi 3-9. doi:10.1093/ndt/gfh1092.
35 Mason D, Schoneich C, Kerwin B. Effect of pH and Light on Aggregation and Conformation of an IgG1 MAb. Mol. Pharm. 9(4) 2012: 774–790; doi:10.1021/mp2004719.
36 Anfinsen CB. Principles That Govern the Folding of Protein Chains. Science 181(4096) 1973: 223–230.
37 Langer T, et al. Chaperonin-Mediated Protein Folding: GroES Binds to One End of the GroEL Cylinder, Which Accommodates the Protein Substrate Within Its Central Cavity. EMBO J. 11(13) 1992: 4757–4765.
38 Wang X, et al. Potential Aggregation Prone Regions in Biotherapeutics: A Survey of Commercial Monoclonal Antibodies. MAbs 1(3) 2009: 254–267.
39 Porter S. Human Immune Response to Recombinant Human Proteins. J. Pharm Sci. 90(1) 2001: 1–11.
40 Schellekens H. Immunogenicity of Therapeutic Proteins: Clinical Implications and Future Prospects. Clin. Therap. 24(11) 2002: 1720–1740.
41 Mok CC, et al. Immunogenicity of Anti-TNF Biologic Agents in the Treatment of Rheumatoid Arthritis. Expert Opin. Biol. Therap. 16(2) 2016: 201–211; doi:10.1517/14712598.2016.1118457.
Corresponding author Xing Wang is president, Lavanya Kolluru and Aarti Madhu were summer interns, and Michael Davies is a senior scientist at Array Bridge Inc., 4320 Forest Park Avenue, Suite 303, Saint Louis, MO 63108; 1-636-284-4212, fax 1-314-932-4038; xing.wang@arraybridge.com