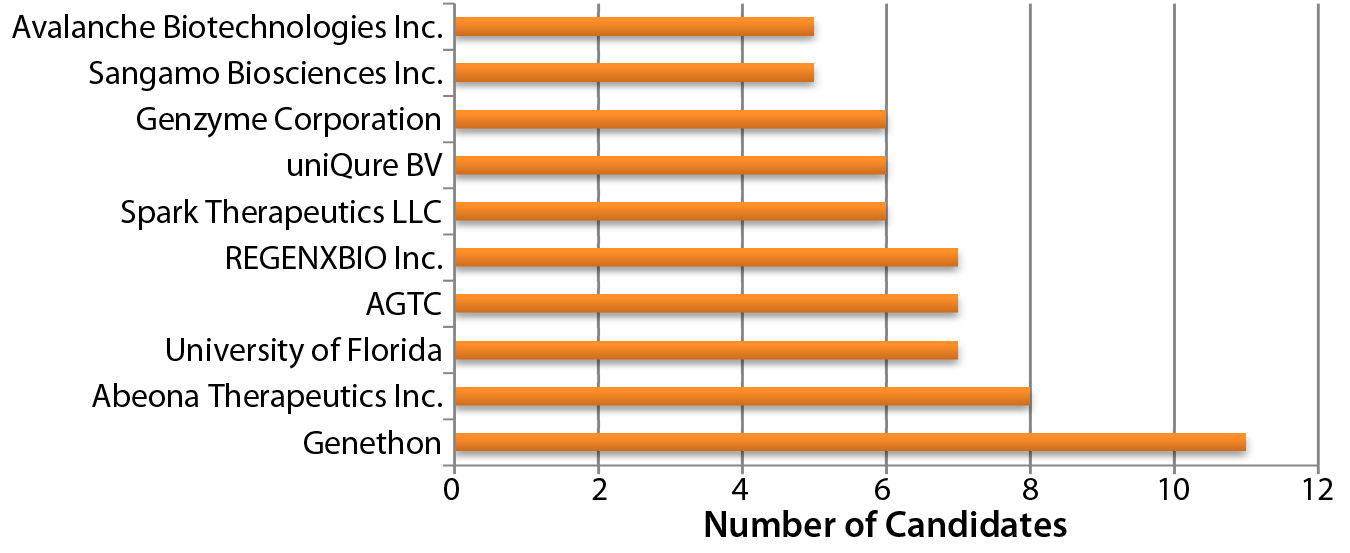
Figure 1: Organizations developing AAV vector candidates
Recent advances in molecular biology are expediting genomic sequencing to underpin precision medicine. Such progress is positioning gene and gene-modified cell therapy on the cusp of an extraordinary revolution in patient care for presently unmet medical needs — and a new therapeutic class that could rival monoclonal antibodies (MAbs) in importance. However, despite substantial strides made in clinical trials, the bioprocessing community is struggling to fulfill growing demands for biomanufacturing capacity to make gene and gene-modified cell therapies — including current good manufacturing practice (CGMP) viral vectors.
Here we review the basic science of gene therapy bioproduction and evaluate critical emerging bioprocess opportunities and challenges. An interrelationship exists between viral-vector upstream and downstream bioprocessing strategies and those for other therapeutic platforms such as chimeric antigen receptor (CAR) T cells and induced pluripotent stem cells (iPSCs).
Fundamental Science Overview
Gene and gene-modified cell therapies treat diseases by delivering therapeutic DNA into a patient’s cells, either in (in vivo) or outside his or her body (ex vivo). It can be used to replace or correct a faulty gene or to encode a therapeutic protein as a treatment. Introducing new DNA often is achieved by viral transduction of target cells with a therapeutic transgene, through either a direct or cell-based gene therapy modality.
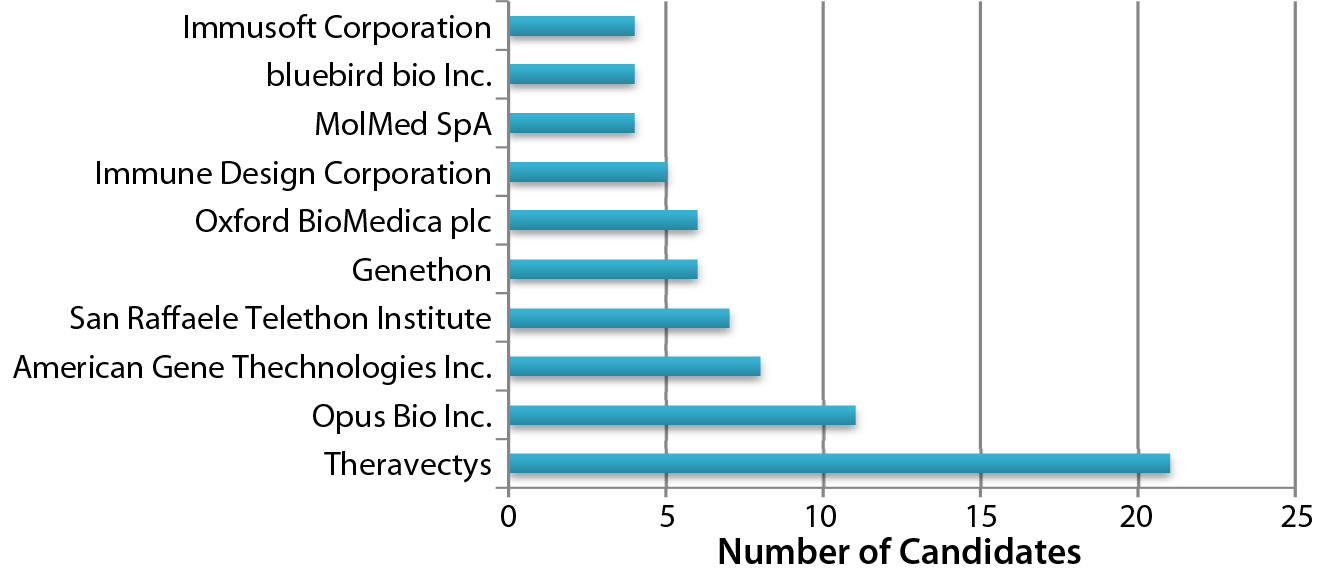
Figure 2: Organizations developing lentiviral vector candidates
The first gene therapy trials were carried out in 1990 treating a four-year old patient who suffered from severe combined immunodeficiency (SCID) (1). Since then, significant progress has been made toward improving the safety and efficiency of viral gene-delivery systems (1). Strategies under investigation include gene replacement and gene knock-out methods as well as classic gene addition (2). Notably, however, most ongoing clinical trials — over 60% of which are directed at cancer — use gene addition as their approach (3).
| Key Definitions |
| Transduction: the process of introducing foreign genetic material into cells using viral vectors |
| Transfection: the process of transferring foreign DNA into host cells (usually refers to nonviral methods) |
| Transformation: genetic alteration of a cell through incorporation of exogenous DNA, which causes transient or stable genetic changes |
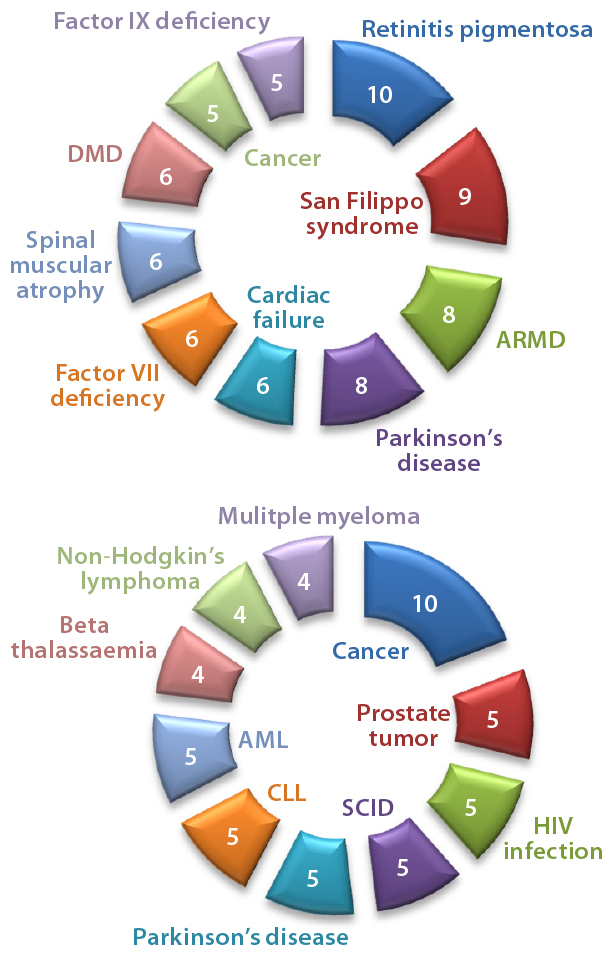
Figure 3: Numbers of drugs and active indications for (top) AAV and (bottom) lentivirus gene therapies
Adenoassociated Virus (AAV) or Lentivirus? Both adenoassociated viruses (AAV) and lentiviruses are currently under development for a number of indications in the gene therapy space. Figure 1 depicts companies that are currently working on AAV-based vectors, and Figure 2 depicts those developing lentiviralbased vectors. Both types are in development for a range of disease conditions (Figure 3).
The first step in both direct and cell-based gene therapy is packaging a therapeutic transgene into a delivery vehicle (e.g., a viral vector), followed by expansion of its host cell lines to produce high-enough vector concentrations. In direct gene delivery, therapeutic transgenes are injected directly into target tissues in vivo; cell-based gene therapy requires isolation and ex vivo cultivation of the target cells before genetic modification of them, followed by expansion and enrichment for readministration into a patient. Figure 4 provides a manufacturing process overview.
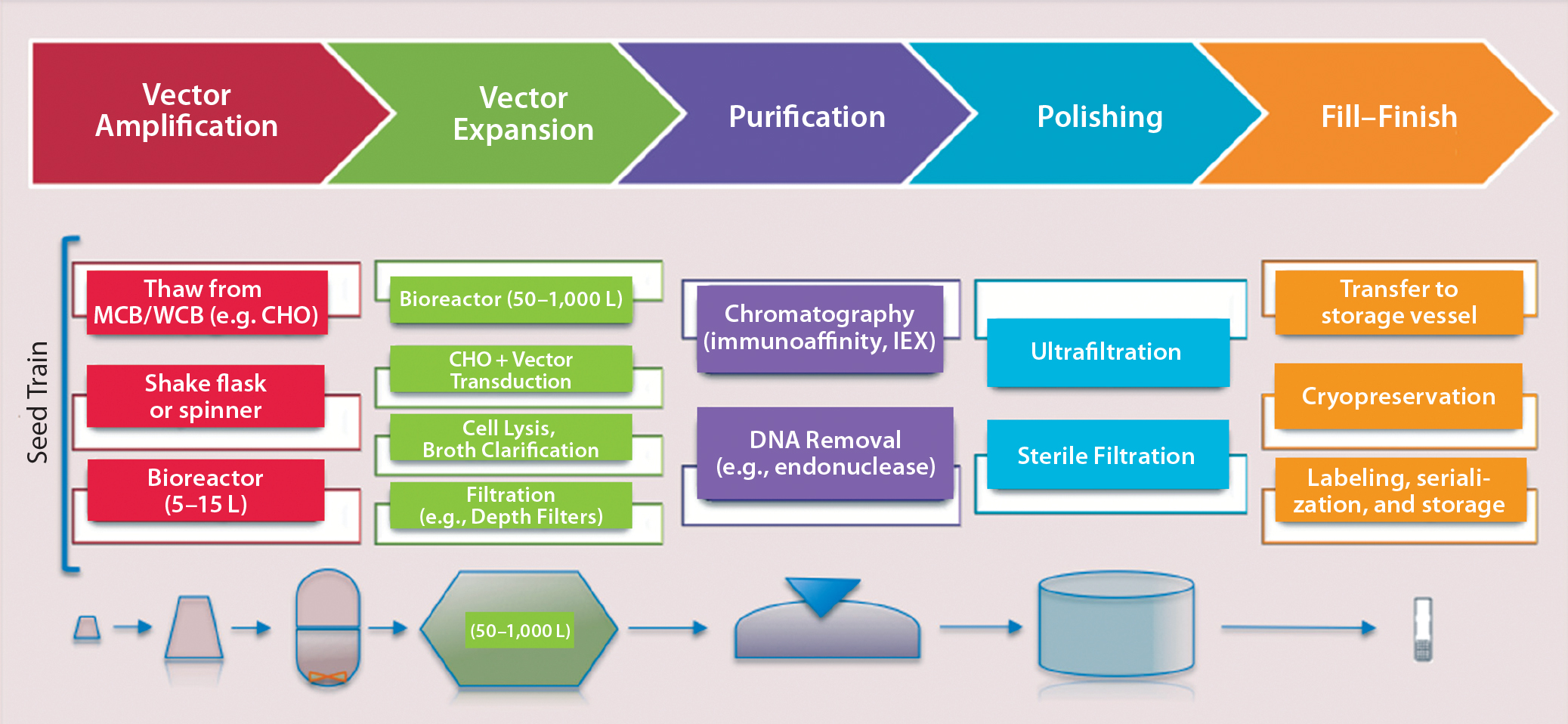
Figure 4: Overview of a typical viral-vector manufacturing process
For improved process economics, batches can be combined to make larger quantities of a final vector (drug) product. Once a target number of subbatches are available with sufficient titers, they can be bulked together in a sterile manufacturing process involving sterilizing filtration with further processing and vialing under aseptic conditions (to achieve a target vector concentration). For certain in vivo gene therapy applications, this could require higher volumetric concentrations (e.g., ≤2,000-fold) than are necessary for ex vivo applications.
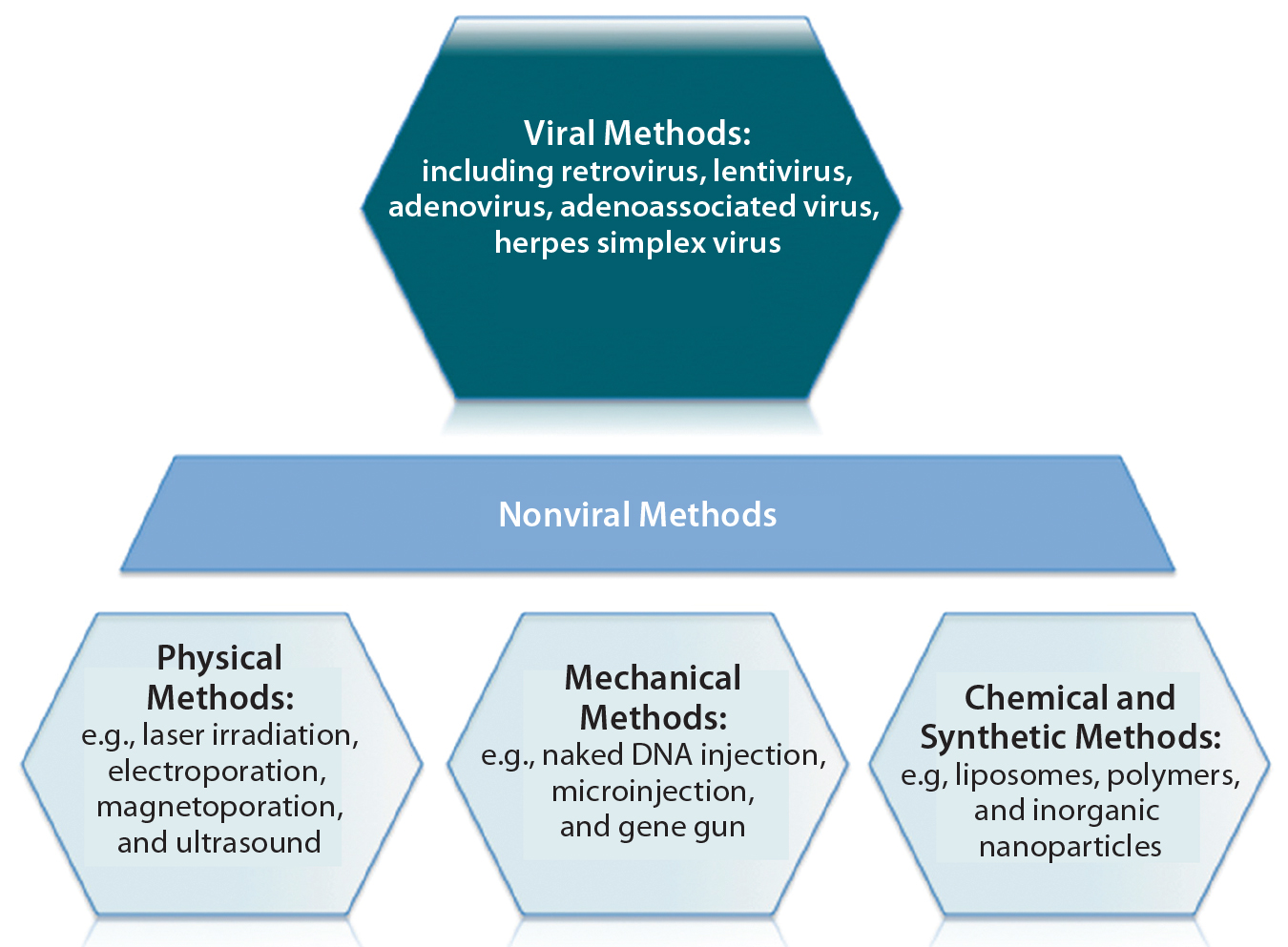
Figure 5: An overview of gene therapy vectors (adapted from An Introduction to Transfection Methods. Amaxa (now Lonza AG) 2007; http://bio.lonza.com/fileadmin/groups/marketing/DMS/downloads/TechRefGuide_AnIntroTransMeth.pdf)
Several types of viruses can be used as vectors to transfer a functional copy of a gene into a patient’s cells (Figure 5). Vectors based on the nonpathogenic AAV human parvovirus are well suited for direct gene therapy (4). AAV vectors are characterized by low immunogenicity, which has made them safe for direct in vivo applications so far (4). Glybera alipogene tiparvovec from uniQure (Amsterdam, The Netherlands), the first commercially available gene therapy in Europe, is based on an AAV1 vector carrying the human lipoprotein lipase (LPL) gene (5). It is used by intramuscular injection for adult patients diagnosed with familial lipoprotein lipase deficiency (LPLD) and suffering from severe or multiple pancreatitis attacks despite dietary fat restrictions.
Viral vectors that can infect dividing and nondividing cells are used for most approaches to cell-based gene therapy. They genetically modify target cells such as T cells or adult cells to induce immune function or pluripotency. Successful application depends on two key principles: identifying an appropriate therapeutic gene and successfully delivering it to the target site while maintaining its efficacious properties (e.g., for gene expression).
Use of a third-generation minimal lentiviral vector that incorporates key safety features is essential. They collectively prevent the reacquisition of replication competence (e.g., codonoptimized gag/pol to minimize homology among vector components and prevent recombination, and selfinactivating long-terminal repeat (LTR) sequences, and to remove all unnecessary sequences and accessory genes (6). Typically a gag/pol packaging construct encodes viral structure proteins (gag) and enzymes (pol), with an additional construct encoding a suitable envelope glycoprotein from a heterologous source, resulting in vector particle “pseudotyping.” That creates a third construct for expression of viral accessory protein Rev along with a fourth vector plasmid encoding the transgene as well as other sequences required for efficient reverse transcription, RNA packaging, and integration (7).
Identifying an appropriate therapeutic gene and maintaining optimum transgene expression presents a considerable challenge in gene therapy development, requiring precise transgene regulation to mitigate undesirable effects (8). Two critical genetic elements regulate transgene expression: promoters and enhancers. Categorized as constitutive or inducible (and functioning either generally or specific to certain tissues), promoters enable either continuous (constitutive) or transient (inducible) transcription of a specific gene (9). Although some such as the melanocyte-specific tyrosinase promoter display robust transcriptional activity and high specificity, other promoters are less effective and require a degree of “enhancement” to bolster their effects (9). That can be achieved through a number of strategies, including elimination of defunct promoter regions (10), duplication of sites exhibiting positive regulatory components (11), combinations of different tissue-specific promoters (9), and using recombinant transcriptional activators (RTAs) to facilitate DNA binding and transactivation domains from a selection of distinctive proteins (12). Once a suitable therapeutic gene is identified, it must be effectively delivered to target cells.
Vector-Based Delivery: Targeted gene delivery involves broadening the tropism of vectors to transduce previously nonpermissive cells or replacing the viral tropism to transduce specific target cell exclusively. Such approaches offer potential advantages of enhanced therapeutic effects and reduced side effects. To deliver a gene therapy to its target site, an appropriate vehicle must be chosen. Vector-based delivery can be broadly categorized as viral or nonviral (Figure 5). Vectors ideally should exhibit a number of key properties:
- tightly regulated expression
- a low adverse-effect profile
- sustained and controllable therapeutic effects
- target-cell specificity
- size capacity.
Plasmid DNAs (pDNAs) serve as nonviral gene delivery vehicles for some gene therapies. With a favorable safety profile, low toxicity, and an ability to deliver larger genetic loads, pDNAs provide an alternative option to viral-mediated approaches that is amenable to a number of physical and mechanical delivery strategies (13). They are, however, limited by “biological barriers” such as renal clearance, endosomal attack, and degradation by serum endonucleases (14). Therefore, about 70% of gene therapy clinical trials to date have involved viral-vector systems instead (14).
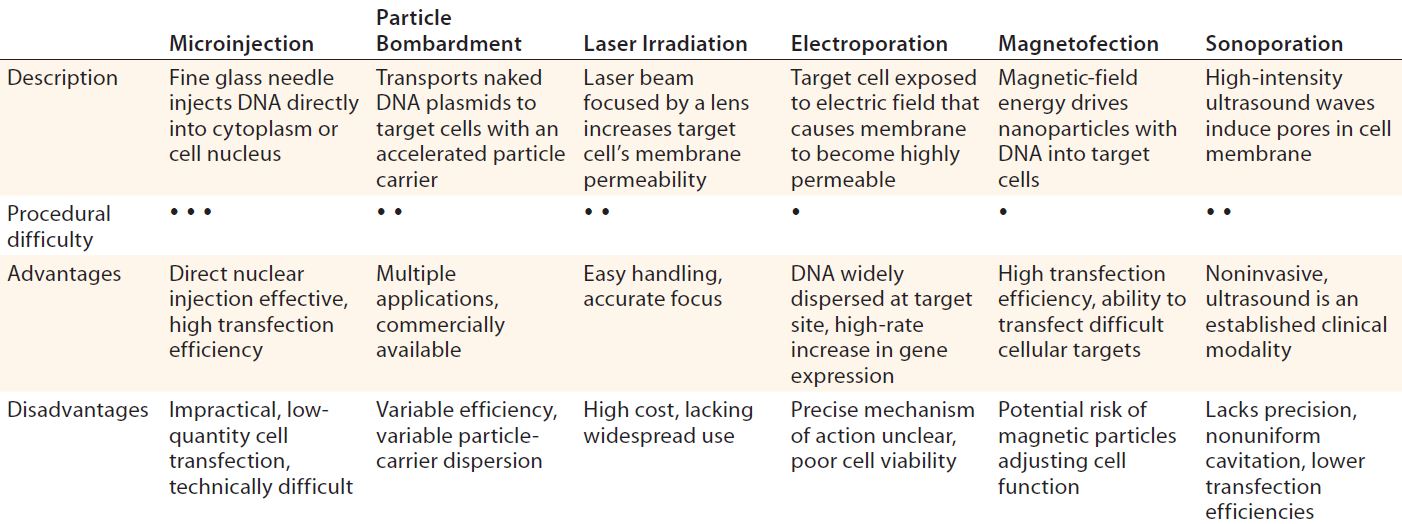
Table 1: Physical and mechanical delivery methods overview (13, 17–19); procedural difficulty scale — high (• • •), moderate (• •), low (•)
Mechanical and Physical Delivery: A number of modalities have been engineered to mechanically and physically deliver gene therapies to their targets. The repository of available techniques includes microinjection, particle bombardment (“gene gun” technology), laser irradiation, electroporation, sonoporation, and magnetofection (Table 1). Such methods attempt to deliver nucleic acids to cells and offer a favorable alternative to vector-based approaches associated with target- and immunogenicity-related complications (15). In cardiology, for example, a number of mechanical/physical options have been adopted specifically for management of cardiovascular disease. They include the use of catheters (for anterograde or retrograde infusion) and direct injection (percutaneous intramyocardial or surgically invasive administration) aided by electrochemical mapping (16).
Microinjection techniques were first devised for gene therapy purposes in 1980 with injection of foreign DNA into both mammalian-cell cytoplasm and native DNA (17). It is common in modern-day practice to produce transgenic mice (18). Despite being conceptually simplistic, the single-cell transfection methodology as a procedure allows transfer of only a few hundred cells, so it has thus been largely replaced by alternative mechanical, physical, and vector-based delivery methods.
Particle bombardment, also known as gene-gun or ballistic gene delivery, uses heavy-metal particles to propel naked DNA plasmids into target cells. This technique has been used cautiously in managing a number of clinical indications such as cancer therapy, genetic vaccination, and wound healing. But it relies on optimal DNA loading of particles, even down to final distribution and delivery timing, among other complex factors (19).
Physical gene delivery approaches include laser irradiation. Focusing an irradiation source such as titanium sapphire or neodymium-doped yttrium aluminum garnet (Nd:YAG) on a specific cell target increases its permeability at the site of impact. That enables any genes present in the surrounding medium to cross cell membranes and enter (19). As a relatively new concept, laser-based approaches lack widespread clinical adoption and require ongoing research.
Electroporation also augments the permeability of cell membranes — in this case by exposing target cells to an electric field. As with laser irradiation, the transient cell-membrane disruption allows genetic material present in surrounding medium to enter through nanometer-size transient aqueous pores (20). A few devices are commercially available for this use, but the specific mechanism of action pertaining to those porous openings is contentious. Again, further research is required for electroporation to progress beyond preclinical studies and into practice (21).
Magnetofection uses a magnetic field to enhance transfection of biodegradable nanoparticles loaded with DNA and an associated transfection component. The external magnetic field drives those particles into target cells. Some favorable outcomes with this technology have been reported (15, 19, 22), highlighting its application to a range of primary cell types with promising efficiency (23).
Finally, ultrasound also has been used to induce pore formation in cell membranes through a method known as sonoporation. High-intensity ultrasonic waves instigate the physical movement of extracellular fluid, leading to direct membrane cavitation or air-bubble collapse and subsequent pore induction (15). Although this method has been successful with a number of cell and tissue types (24), it is limited by imprecise energy localization and indiscriminate cavitation resulting in lower transfection efficiencies. If those limitations can be addressed successfully, sonoporation may provide a viable means of cellular transfection.
For mechanical and physical approaches to offer viable therapeutic options, a number of challenges relating to both cell membranes and the intracellular environment must be overcome in addition to the technique-specific refinements mentioned above. Several technologies have demonstrated efficacy in generating cell-membrane pores; however, the precise characteristics of those pores (e.g., diameter and duration) have yet be well controlled. Ongoing research efforts must focus on establishing critical quality attributes (CQAs), which would help researchers optimize their gene-delivery applications and clinical efficacy.
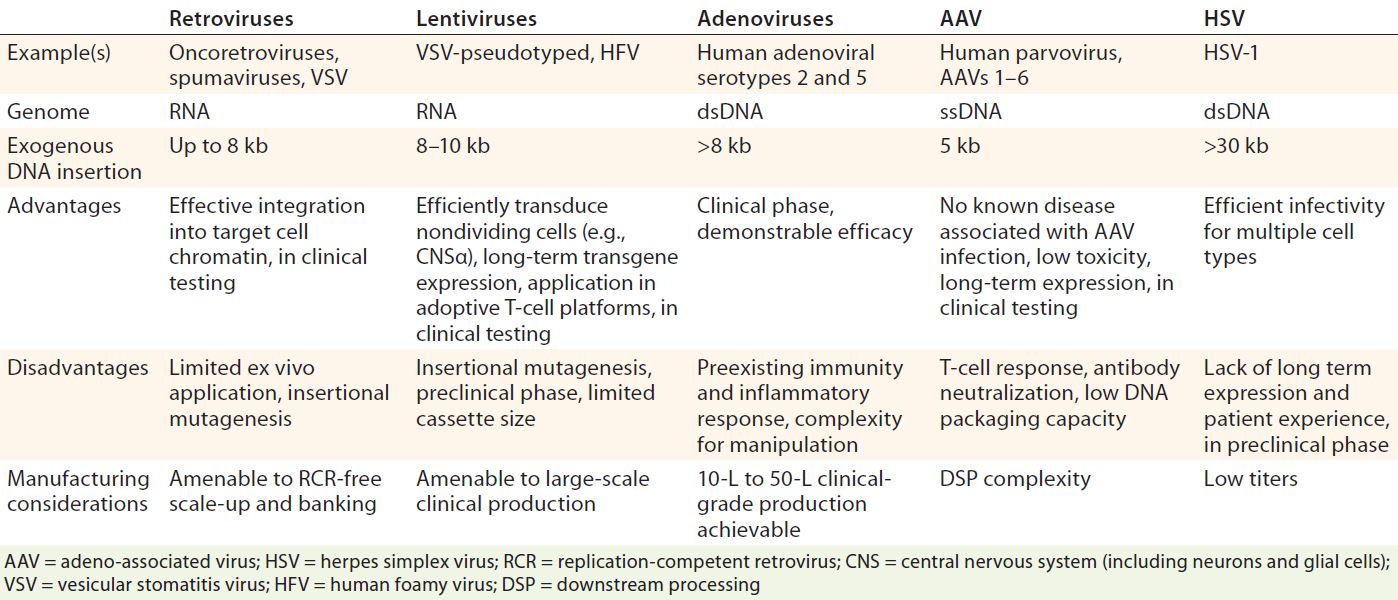
Table 2: Viral vectors overview (14, 23, 25, 31)
Viral Vectors: Viruses are naturally abundant, optimally characterized delivery vehicles for transfer of genetic information, so they have been the focus of significant clinical research efforts (see Table 2). The natural life cycle of a virus can be divided into two distinct phases: infection and replication. The former causes uptake of viral genomes by host cells and leads to subsequent gene expression (25). That is exploited in gene therapy, for which a viral genome is replaced with an encapsulated therapeutic gene cassette containing a modified genome, which is consequently introduced to target cells (25). For efficacious disease management, the amounts of therapeutic genetic material entering target cells must be precisely controlled in accordance with properties exhibited by a given disease. Treatment of genetic disorders may require long-term rather than short-term gene expression. Careful selection (and production) of viral vectors is essential to achieving such effects.
Viral Manufacturing
To manufacture viral vectors, two approaches generally are adopted for culturing purposes: scale-out of adherent-cell systems based on 2D planar technologies and scale-up of 3D suspension cell cultures in stirred-tank bioreactors.
For large-scale manufacturing, the conventional laboratory processes of adherent-cell systems (e.g., T-flasks, roller bottles, hollow-fiber technologies, and multilayer flasks) pose considerable challenges in scaling up (4). Although estimates citing the exact quantities of viral vectors required to meet clinical demand vary (26), developers are increasingly moving away from small-scale operations and choosing to operate larger-scale apparatus such as stainless steel or single-use bioreactor culture systems (4, 27). Viral sensitivity and stability must be considered diligently at the forefront of scale-up planning. Factors such as shear forces, pH conditions, and temperature become increasingly important as yields increase incrementally (28, 29).
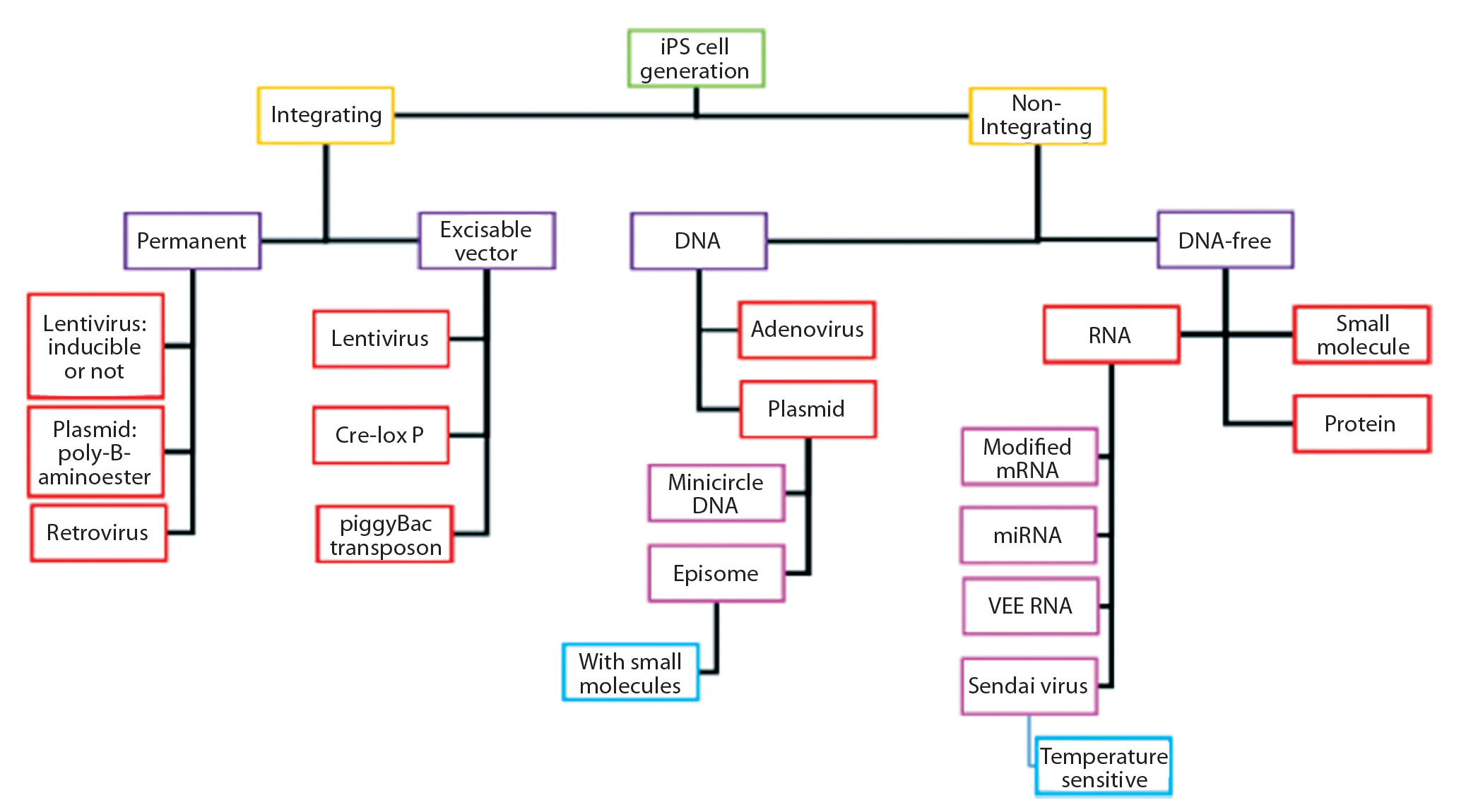
Figure 6: Delivery methods and technologies for iPSC manufacturing (29)
Lentiviral vectors emerged in the 1990s as a safe alternative to γ-retroviral vectors. Lentiviruses are rapidly gaining traction as important tools for gene and gene-modified cell therapy because of their ability to insert genes into dividing and nondividing cells. Application of these vectors in production of induced pluripotent stem cells (iPSCs) (Figure 6) (27, 30) as well as direct gene therapeutics or activating agents for immunotherapy make them a cornerstone technology in the advancement of regenerative medicine. Because of the limitations of conventional laboratory-based culture methods and an increasing demand for lentiviral vectors, newer, larger-scale production methods are increasingly used. They include disposable fixed-bed bioreactors, closed-system hollow-fiber bioreactors, and single-use bioreactors. Vector titers in hollow-fiber bioreactors have been demonstrated as comparable to those attained with flask or Nunc Cell Factory tray cultures: 1.7 × 108 viral genomes per milliliter (vg/mL) to 2.6 × 108 vg/mL (27). However, suspension-adapted production cell lines cultured in large-scale bioreactors are more amenable to therapeutic product expression because of their enhanced scalability and elimination of detachment agents, which facilitates regulatory compliance.
Adenoviral vectors can be produced by human embryonic kidney (HEK293) and human embryonic retinal (PER.C6) cell lines (31), both of which also can be used for lentiviral vector production. Driven by increasing clinical demands, manufacturing technologies for adenoviral vector production also have advanced to help companies increase their total production yields. In addition to fixed-bed reactors, 2D planar technologies systems, and parallel processing of multiple units, bioreactors containing microcarrier beads have been used (32). However, the industry appears to favor cells adapted to suspension cultures (e.g., HEK293), which can successfully grow in serum-free media and thus provide a heightened safety profile and reduced cost of goods (CoG) (33). Serum-free suspension cultures are operationally straightforward and amenable to industrial scale-up into stirred-tank bioreactors, allowing for 1,000-L to 10,000-L production capacities (32, 34, 35).
For AAV manufacturing, reported transient transfection allows obtainable yields in the region of 1015– 1016 vg, which is generally sufficient for AAV-based gene therapy regimes (36). However, current proprietary transfection technologies may well exceed those reported numbers. Doing so may require expansion to meet broader clinical indications or higher dose requirements. Approaches such as the baculovirus expression vector system (BEVS) in use at uniQure (Amsterdam, The Netherlands) for manufacturing alipogene tiparvovec may provide suitable options (4). Developed by Kotin et al. (37), the BEVS uses Spodoptera frugiperda (Sf9) cells infected with recombinant baculoviruses containing the vector genome, helper genes, and capsids to generate yields of 1014–1016 viral genomes per liter in 100-L to 200-L bioreactors (4, 38). Greater yields are expected to come with progressive scale-up (39). Other scalable systems include cell lines based on the Henrietta Lacks tumor (HeLa) and baby hamster kidney (BHK-21) cells for adenovirus serotype 5 (Ad5) and herpes simplex virus 1 (HSV-1) production (4, 40, 41). The BHK-21 system has attained yields up to 1014 vg/L using 10–L rocking-motion disposable bioreactors (4, 40, 41).
For downstream processing (particularly viral purification), a number of strategies are used to select for target product(s) and eliminate unwanted contaminants. Careful consideration must be given to each distinct type of viral vector, however, because of differences in their biophysicochemical properties and surface characteristics (32). A number of strategies focus on viral size discrimination (29) or electrical charge. With an overall industry trend to move away from traditional purification processes such as ultracentrifugation because of their limitations relating to scalability and labor intensity, chromatographic technologies more amenable to scaleup are gaining popularity. Selective binding chemistries relating to virus-particle surfaces can be influenced through environmental adjustments such as suspension pH or adding charged exchange media and membranes (29, 42).
As with all manufacturing processes associated with therapeutics intended for human use, quality control (QC) and release testing are essential activities to ensure product safety, purity, and potency (43). The structural complexity of viral vectors makes QC testing challenging. Novel analytical methods systematically address each individual viral-vector–based therapy. Certain measures can be taken to improve the robustness and quality of manufacturing processes. They include working toward closed systems (to reduce the possibility of operator processing errors, minimize open manual-handling steps, streamline process flows, and reduce opportunities for product-contamination), for example with commercially available good manufacturing practice (GMP) compliant culture bags, connectors, and accessories; standardized input cellular material; and clearly defined ancillary reagents (44). Process control in addition to simplification of multistep manufacturing processes (e.g., through automation and process simplification) will reduce variability, facilitate QC and release testing, and help companies work toward attainment of CGMP-compliant products (45).
Clinical Applications and Bioprocessing Strategies
Fundamentally, gene therapy should exceed the efficacy of existing therapeutic approaches and provide therapeutic strategies for unmet medical needs. However, it is not a stand-alone technology. Native viral vectors can be therapeutics themselves, but they also function as a critical input to manufacturing rapidly emerging therapeutic platforms, including CAR-T and other forms of gene-modified whole-cell therapies. So predicting future biomanufacturing demands for viral vectors is a challenge. At a minimum, however, the bioprocess community must prepare itself to meet the needs of both “pure” gene therapies and adoptive immunotherapies in development (Figures 2 and 3). Ultimately this will include the expanding repertoire of advanced technologies such as clustered regularly interspaced short palindromic repeats (CRISPR), CRISPR-associated protein 9 (Cas9), and zinc-finger nucleases (ZFNs) (46) along with the new clinical indications and markets that they address. To ensure effective clinical translation and commercialization, the bioprocessing community must devote intense effort to scalability of vector manufacturing. Solutions may include process scheduling in flexible facility management, the application of process analytical technologies (PATs) to maximize process control, and support innovation in QA technologies to reduce their impact on CoG.
Acknowledgments
We express sincere thanks to the following organizations that have contributed to the CASMI Translational Stem Cell Consortium (CTSCC) as funding and events partners, without whom the consortium and the benefits it will bring to stem cell translation would be constrained: GE Healthcare, the Center for Commercialization of Regenerative Medicine (CCRM), Sartorius Stedim Biotech (formerly TAP Biosystems), Lonza, the California Institute for Regenerative Medicine (CIRM), the Strategies for Engineered Negligible Senescence (SENS) Research Foundation, UK Cell Therapy Catapult, NIH Centre for Regenerative Medicine, the New York Stem Cell Foundation (NYSCF), ThermoFisher Scientific, Eisai, Medipost (US), Medipost (Korea), Celgene, Roche and Oxford BioMedica (UK) Ltd. David Brindley gratefully acknowledges personal funding from the Oxford Musculoskeletal National Institute for Health Research (NIHR), the Saïd Foundation, and the SENS Research Foundation. David Pettit acknowledges support from the SENS Research Foundation; he and James Smith gratefully acknowledge support from the CASMI Translational Stem Cell Consortium (CTSCC). And Smith also gratefully acknowledges support from the UK Medical Research Council and Professor Andrew Carr at the Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences in the University of Oxford’s medical services division.
Conflicts of Interest
This text represents the authors’ individual opinions and may not necessarily represent the viewpoints of their employers. Brindley is a stockholder in Translation Ventures Ltd. (Charlbury, UK) and IP Asset Ventures Ltd. (Oxford, UK), companies that among other services provide cell therapy biomanufacturing, regulatory, and financial advice to pharmaceutical clients. Smith is a consultant with IP Asset Ventures Ltd. Brindley also is subject to the CFA Institute’s codes, standards, and guidelines, so he must stress that this piece is provided for academic interest only and must not be construed in any way as an investment recommendation. Finally, at time of publication, he and the organizations with which he is affiliated may or may not have agreed to and/or have pending funding commitments from organizations named herein.
References
1 Verma IM, Somia N. Gene Therapy – Promises, Problems and Prospects. Nature 389(6648) 1997: 239–242.
2 Somia N, Verma IM. Gene Therapy: Trials and Tribulations. Nat. Rev. Genetics 1(2) 2000: 91–99.
3 Collins M, Thrasher A. Gene Therapy: Progress and Predictions. Proc. Biolog. Sci. Royal Soc. 282(1821) 2015; doi:10.1098/rspb.2014.3003.
4 Van der Loo JC, Wright JF. Progress and Challenges in Viral Vector Manufacturing. Hum. Molec. Genetics 2015.
5 Moran N. First Gene Therapy Approved. Nat. Biotechnol. 30(12) 2012: 1153– 1153.
6 Dull T, et al. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 72(11) 1998: 8463–8471.
7 Kim VN, et al. Minimal Requirement for a Lentivirus Vector Based on Human Immunodeficiency Virus Type. J. Virol. 72(1) 1998: 811–816.
8 Das SK, et al. Gene Therapies for Cancer: Strategies, Challenges and Successes. J. Cellul. Physiol. 230(2) 2015: 259–271.
9 Nettelbeck DM, Jérôme V, Müller R. Gene Therapy: Designer Promoters for Tumour Targeting. Trends Genet. 16(4) 2000: 174–181.
10 Siders WM, et al. Melanoma-Specific Cytotoxicity Induced By a Tyrosinase Promoter-Enhancer/Herpes Simplex Virus Thymidine Kinase Adenovirus. Cancer Gene Ther. 5(5) 1998: 281–291.
11 Pang S, et al. Identification of a Positive Regulatory Element Responsible for Tissue-Specific Expression of Prostate-Specific Antigen. Cancer Res. 57(3) 1997: 495–499.
12 Nettelbeck D, Jerome V, Müller R. A Strategy for Enhancing the Transcriptional Activity of Weak Cell Type-Specific Promoters. Gene Ther. 5(12) 1998: 1656–1664.
13 Bathula SR, Huang L. Gene Therapy with Plasmid DNA. Burger’s Medicinal Chemistry and Drug Discovery. John Wiley & Sons, Inc.: Hoboken, NJ, 2010; doi:10.1002/0471266949.bmc073.pub2.
14 Yin H, et al. Non-Viral Vectors for Gene-Based Therapy. Nat. Rev. Genet. 15(8) 2014: 541–555.
15 Mellott AJ, Forrest ML, Detamore MS. Physical Non-Viral Gene Delivery Methods for Tissue Engineering. Ann. Biomed. Eng. 41(3) 2013: 446–468.
16 Rincon MY, Vandendriessche T, Chuah MK. Gene Therapy for Cardiovascular Disease: Advances in Vector Development, Targeting, and Delivery for Clinical Translation. Cardiovasc. Res. 108(1) 2015: 4–20.
17 Capecchi MR. High Efficiency Transformation By Direct Microinjection of DNA into Cultured Mammalian Cells. Cell 22(2) 1980: 479–488.
18 Gordon JW, Ruddle FH. Gene Transfer into Mouse Embryos: Production of Transgenic Mice By Pronuclear Injection. Meth. Enzymol. 101(28) 1983: 411–433.
19 Mehier-Humbert S, Guy RH. Physical Methods for Gene Transfer: Improving the Kinetics of Gene Delivery into Cells. Adv. Drug Deliv. Rev. 57(5) 2005: 733–753.
20 Venslauskas MS, Šatkauskas S. Mechanisms of Transfer of Bioactive Molecules Through the Cell Membrane By Electroporation. Eur. Biophys. J. 44(5) 2015: 277–289.
21 Dean DA. Cell-Specific Targeting Strategies for Electroporation-Mediated Gene Delivery in Cells and Animals. J. Membrane Biol. 246(10) 2013: 737–744.
22 Plank C, et al. The Magnetofection Method: Using Magnetic Force to Enhance Gene Delivery. Biolog. Chem. 384(5) 2003: 737–747.
23 Buerli T, et al. Efficient Transfection of DNA or shRNA Vectors into Neurons Using Magnetofection. Nat. Protocols 2(12) 2007: 3090–3101.
24 Newman C, Bettinger T. Gene Therapy Progress and Prospects: Ultrasound for Gene Transfer. Gene Ther. 14(6) 2007: 465–475.
25 Kay MA, Glorioso JC, Naldini L. Viral Vectors for Gene Therapy: The Art of Turning Infectious Agents into Vehicles of Therapeutics. Nat. Med. 7(1) 2001: 33–40.
26 Lyddiatt A, O’Sullivan DA. Biochemical Recovery and Purification of Gene Therapy Vectors. Curr. Opin. Biotechnol. 9(2) 1998: 177–185.
27 Sheu J, et al. Large-Scale Production of Lentiviral Vector in a Closed System Hollow Fiber Bioreactor. Molec. Ther. Meth. Clin. Dev. 2, 2015: 15020.
28 Walther W, Stein U. Viral Vectors for Gene Transfer. Drugs 60(2) 2000: 249–271.
29 Morenweiser R. Downstream Processing of Viral Vectors and Vaccines. Gene Ther. 12, 2005: S103–S110.
30 Silva M, et al. Generating iPSCs: Translating Cell Reprogramming Science into Scalable and Robust Biomanufacturing Strategies. Cell Stem Cell 16(1) 2015: 13–17.
31 Aurisicchio L, Ciliberto G. Genetic Cancer Vaccines: Current Status and Perspectives. Exp. Opin. Biolog. Ther. 12(8) 2012: 1043–1058.
32 Kallel H, Kamen AA. Large-Scale Adenovirus and Poxvirus-Vectored Vaccine Manufacturing to Enable Clinical Trials. Biotechnol. J. 10(5) 2015: 741–747.
33 Garnier A, et al. Scale-Up of the Adenovirus Expression System for the Production of Recombinant Protein in Human 293S Cells. Cell Culture Engineering IV. Buckland BC, Ed. Springer Science and Business Media: Rotterdam, The Netherlands, 1994; 145–155.
34 Xie L, et al. Large-Scale Propagation of a Replication-Defective Adenovirus Vector in Stirred-Tank Bioreactor PER.C6™ Cell Culture Under Sparging Conditions. Biotechnol. Bioeng. 83(1) 2003: 45–52.
35 Brindley DA, et al. Peak Serum: Implications of Serum Supply for Cell Therapy Manufacturing. Regen. Med. 7(1) 2012: 7–13.
36 Wright F. Transient Transfection Methods for Clinical Adeno-Associated Viral Vector Production. Hum. Gene Ther. 20(7) 2009: 698–706.
37 Urabe M, Ding C, Kotin RM. Insect Cells As a Factory to Produce AdenoAssociated Virus Type 2 Vectors. Hum. Gene Ther. 13(16) 2002: 1935–1943.
38 Cecchini S, Virag T, Kotin RM. Reproducible High Yields of Recombinant Adeno-Associated Virus Produced Using Invertebrate Cells in 0.02-to 200-Liter Cultures. Hum. Gene Ther. 22(8) 2011: 1021– 1030.
39 Wright JF. Adeno-Associated Viral Vector Manufacturing: Keeping Pace with Accelerating Clinical Development. Hum. Gene Ther. 22(8) 2011: 913–915.
40 Thomas DL, et al. Scalable Recombinant Adeno-Associated Virus Production Using Recombinant Herpes Simplex Virus Type 1 Coinfection of Suspension-Adapted Mammalian Cells. Hum. Gene Ther. 20(8) 2009: 861–870.
41 Thorne BA, Takeya RK, Peluso RW. Manufacturing Recombinant Adeno-Associated Viral Vectors from Producer Cell Clones. Hum. Gene Ther. 20(7) 2009: 707– 714.
42 Specht R, et al. Densonucleosis Virus Purification By Ion Exchange Membranes. Biotechnol. Bioeng. 88(4) 2004: 465–473.
43 Brindley DA, et al. The Potential Application of Real-Time Release Testing for the Biomanufacture of Autologous Cell-Based Immunotherapies. BioProcess Int. 13(4) 2015: S34–S43.
44 Kaiser AD, et al. Towards a Commercial Process for the Manufacture of Genetically Modified T Cells for Therapy. Cancer Gene Ther. 22(2) 2015: 72–78.
45 Brindley DA, Wall I, Bure K. Automation of Cell Therapy Biomanufacturing. BioProcess Int. 11(3) 2013: S18–S25.
46 Baek K, et al. Gene Transfection for Stem Cell Therapy. Curr. Stem Cell Rep. 2016: 1–10.
Corresponding author David Pettitt is an academic surgeon and a doctoral researcher at the Oxford–UCL Centre for the Advancement of Sustainable Medical Innovation (CASMI) and in the medical sciences division’s department of pediatrics at the University of Oxford, UK, and an associate at IP Asset Ventures Ltd.; david.pettit@hertford.ox.ac.uk. James Smith is a CASMI research associate and doctoral researcher in the University of Oxford’s Nuffield Department of Orthopedics, Rheumatology and Musculoskeletal Sciences (Oxford, UK), and an associate at IP Asset Ventures, Ltd. Maya Fuerstenau-Sharp is a field marketing manager, and Kim Bure is director of regenerative medicine at Sartorius Stedim (Göttingen, Germany). Georg Holländer is an action research professor in the medical sciences division’s department of pediatrics at the University of Oxford and the Weatherall Institute of Molecular Medicine at John Radcliffe Hospital (Oxford, UK). Paul Predki and Andrew Slade are with IP Asset Ventures Ltd. (Oxford, UK). Peter Jones is head of manufacturing development at Oxford BioMedica (Oxford, UK). Kyriacos Mitrophanous is chief scientific officer at Oxford BioMedica. And David Brindley is founder and academic director of CASMI’s Translational Stem Cell Consortium, senior healthcare translation research fellow in the medical sciences division’s department of paediatrics at the University of Oxford, Cooksey-Saïd fellow in healthcare translation at the University of Oxford’s Saïd Business School, honorary senior research associate at the University College London School of Pharmacy’s Centre for Behavioral Medicine (London, UK), a research fellow in cell therapy commercialization at the Harvard Stem Cell Institute (Cambridge, MA), and regenerative medicine regulation and risk management lead at the USCF-Stanford Center of Excellence in Regulatory Science and Innovation (CERSI) in Stanford, CA.