Live whole-cell bacterial products have been used as vaccines for many years, and there are currently three such products licensed on the market. Over recent years, however, interest has renewed in this type of product as a delivery system for novel recombinant therapies and vaccines. A number of different organisms have been proposed, such as Escherichia coli and Salmonella species, which might have applicability for such applications.
Vaccine applications tend to relate to the potential for low-cost orally delivered products against a range of infectious diseases. Injectible products are more focused on delivery of specific therapies such as cancer vaccines. Use of recombinant expression creates the opportunity for development of platform delivery systems for a wide range of therapeutic agents. This suggests the possibility of developing platform manufacturing processes that could be used for multiple products, including those intended for early stage clinical trials. This goes against the conventional expectation of product-dedicated facilities and in turn requires a new and different approach for the manufacture of these products both for clinical-trial and market applications.
PRODUCT FOCUS: V
In 2007, our company was approached to manufacture a recombinant Listeria monocytogenes vaccine to be used against cervical cancers. The customer required enough product for phase 2 clinical trials in the United States. This product was intended to be delivered by intratumoral injections, so it required a process that would be validated as monoseptic. The Cobra Biomanufacturing process science group developed a new process to that end based on the use of sterile disposable technologies at all stages of manufacture. The project encompassed process and systems design, fabrication, and validation followed by engineering and manufacturing runs before release of clinical-grade material. The project was achieved within nine months, and the product has now been approved for clinical trials by the US Food and Drug Administration. More information of this specific product is can be obtained from the website of the customer, Advaxis Inc. (www.advaxis.com).
Downstream OptionsAt first, the production of a live whole-cell therapeutic product seems to be a simple process. Cells are grown and recovered, then their fermentation medium is replaced with a formulation buffer. Traditionally, this has been performed by growing cells in shake flasks or fermentors, then recovering cells by centrifugation, and then washing and resuspending them in a formulation buffer (Figure 1).

Figure 1:
The first requirement in production of live-bacterial material intended for clinical use is to develop a process that meets the regulatory requirements for monoseptic products. In the European Union, supporting validation studies must be performed to support the claim of monosepsis for all clinical materials. The second requirement is for a process to be capable of generating material that meets required specifications regarding product activity and contaminants. Finally, the process must be scalable with the intrinsic nature of the product retained.
The existing laboratory-scale process for this product was based on performing cell culture in shake flasks (multiple 250-mL units). Cells were recovered through batch centrifugation, and the product was resuspended within the centrifuge pots to generate a final formulated product. Although that process was satisfactory for production of material for initial potency and safety studies, it was thought to be far from ideal for producing larger quantities of material for clinical studies. The main issue is the ability to maintain (and assure maintenance) of monoseptic conditions throughout manufacture. Three options for an alternative production process were proposed:
-
Perform the existing process in grade A/B environments
-
Develop a process around the use of isolator technologies
-
Develop a process based on the use of cross-flow membrane systems (rather than centrifugation) and sterile disposable components.
The first of those options was considered in depth but ultimately rejected for three main reasons. The first was the space limitation of existing grade A/B environments. The second reason was the requirement for material and product to be taken in and out of a critical environment on multiple occasions, making the maintenance of monosepsis highly operator-dependent, with a high risk of potential failure. The combination of those two factors raised serious concerns over the validation that would be required to achieve the level of surety essential for the release of clinical material. Finally, there was a clear limitation to the scale at which such a process could be performed.
The second option of performing all open operations of a centrifugation-based process within thin-film isolators was also examined. Although such units provide high levels of surety for monosepsis, our group had significant concerns about transferring materials in and out of the isolator environments. In addition, isolator gassing regimes require containers to be exposed not only to high levels of oxidizing agents, but also to elevated temperature for extended periods. So the product’s sensitivity to both the gas and temperature rendered this option unviable.
The remaining option was to perform cell harvest and product formulation with a sterilized cross-flow, tangential-flow filtration (TFF) unit linked into a system of sterile single-use components. We believed this to be the most feasible way forward, and initial agreement from the customer was obtained for the approach. Membrane systems have been used for a number of years in cell concentration operations, but these are predominantly for nonsterile applications without the need to maintain cell viability. Previous experience has shown variable success in the concentration of bacterial cells; because this process was relatively low in volume and cell density, however, we felt that there was a reasonable chance of success.
Testing Filtration Methods: Initial studies were necessary for determining whether it was feasible to recover cells using a membrane-based approach and assessing different pore sizes and membrane configurations. The key challenge would be showing that cells could be concentrated with their viability retained and that the operation could be performed in an acceptable time. We initially assessed hollow-fiber and tangential-flow membranes, both in ultra- and microfiltration formats (Figure 2).
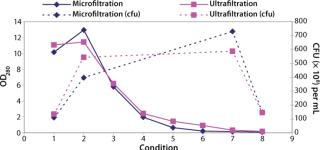
We found that a conventional TFF system gave comparable results to the hollow-fiber systems. But because TFF membranes were unavailable in presterilized formats, they would have required in-situ or autoclaved steam sterilization, a process that would then need validation. We considered procedures for validating such operations to be unproven, so the TFF option was rejected.
Our conclusion from these studies was that a hollow-fiber microfiltration process gave the best results in performing the required concentration and product formulation operations while retaining cell viability. Furthermore, the availability of presanitized hollow-fiber filter units circumvented the need for cleaning validation studies. This gave us confidence in the approach for developing a process that would subsequently fit within a clinical-grade manufacturing environment.
Production OptionsThe second area in the process we addressed was the cell culture itself. Previously this operation had been performed in development programs using shake flasks or a basic batch-fermentation protocol in a 5-L stirred-tank reactor. The available reactors were laboratory units that were not designed for clinical production and thus would require validated sterilization procedures, inlet and outlet connections, and associated control and data logging systems. Procurement of new units would have required significant design input and capital investment, as well as extending timelines to deliver on the project. Therefore, we did not support the use of this type of bioreactor as an option for production of clinical material.
An alternative approach was assessed based on the requirement for a low biomass culture (1×1010 cfu = OD 7–8) for the project and low product volumes of ~5 L of material. Because of these overall requirements, we thought it was possible to move the process to an existing disposable Biostat CultiBag bioreactor from Sartorius Stedim Biotech (www.sartorius-stedim.com), which is known to provide relatively high oxygen transfer (KLa) values for a mammalian-cell reactor system.
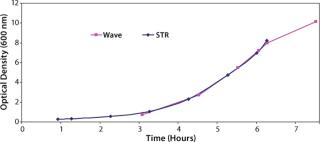
Additionally we expected that the KLa values could be increased if the system was operated at a low fill volume, so we performed a study with the reactor filled to 20% of its full volume rather than the standard value of 50%. Data obtained (Figure 3) showed that it was possible to perform comparable cell growth to that performed in a stirred-tank system, so this would be suitable for the intended application. Having addressed how to grow the bulk cell culture, we had to determine how an initial cell build should be performed to meet the requirement for maintenance of monosepsis throughout the process.
To address that, studies were performed to simplify the inoculum train. Initially we focused on the minimal volume of inoculum that could be used to perform the culture and still achieve acceptable and reliable growth levels and process times, accounting for the fact that cell growth lag time would be increased. Our studies indicated that culture volumes as low as 10 mL would work for this process (Figure 4), and the next challenge was to identify how this initial cell build could be performed as a monoseptic operation.

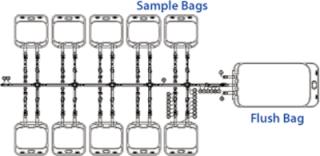
The initial cell build in production of any clinical product requires culturing of cells from a single vial of a previously laid-down and tested cell bank, usually generated in the form of master and working cell banks (MCB and WCB). Regulatory requirements for generation of cell banks currently do not require validated monosepsis. Normal practice would be to lay down up to 200 1-mL vials from shake flask cultures. However, by increasing the volume of our working cell bank vials to 10 mL and filling directly into a sterile bag system (which had tubing outlets that could be directly welded to the cell culture bags), we were confident that our product’s regulatory requirement for monosepsis would be met. To achieve this, we developed a custom bag arrangement by which 10 mL of cell culture could be filled into 60-mL bags. Those bags were manufactured in a manifold arrangement (Figure 5), which could be directly filled from shake flasks and sealed off using tube welders, then frozen as individual “vials.”
From the results obtained from our studies, we believed we had a platform for development of a process that could be performed as a monoseptic operation within GMP. This process is outlined in Figure 6. Our next challenge was take these individual components and build up a working process.

Fluid Train Sterility: The first stage of this work required development of a sterile fluid train that would allow the entire process to be performed as a closed operation. There were a number of basic requirements for this:
-
Link directly to the CultiBag cell culture system
-
Allow the required cell concentration and washing to be performed using a cross-flow membrane system
-
Provide sterile buffer for cell washing and formulation
-
Allow sample collection
-
Provide a sterile barrier for operations including collection of waste solutions such as permeates from the hollow fibers.
We developed an outline system design as a basis for ongoing discussion with our suppliers (Figure 7). This design was based on using welded connections or sterile connectors to bring together a number of units for introduction of the cell culture and washing and formulation buffers, as well as collection and dilution of the final product and removal of required in-process samples.
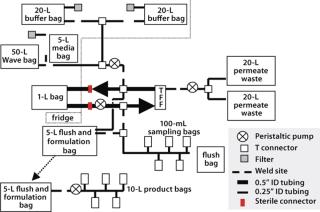
One critical requirement was introduction of sterile liquids (washing and formulation buffers) into the process stream. Regulators expect filters in aseptic filtration operations to be integrity tested before and after use. With irradiated single-use systems (in which filters are normally welded to feed bags) this creates a number of challenges, and the tubing arrangements required to perform such testing procedures are complex and may compromise system sterility. So we took an alternative approach, in which a double 0.2-µm filtration system was used without any preuse integrity testing. Our logic for this approach assumes that the reasoning behind the regulatory requirement is the potential for filters to give false test results at the postuse integrity stage because of blockage. So we prevented such blockage with a double-filtration process, in which all material going on to the second filter will have been 0.2-µm filtered. This justifies performing only postuse integrity testing.
Connection to the CultiBag cell culture system was relatively straightforward and could be achieved by tube-welding the bag outlet to the disposable manufacturing system itself. This did, however, require custom bags with sufficient tube lengths to weld to and provide physical access to the tube-and-bag assemblies. The need to perform such welds (and the tubing lengths required to perform them) would remain a significant challenge throughout the program.
Culture Harvest: Our next decision involved choosing between a membrane-system format that would be prewelded into a tubing system and one that could be connected by sterile connectors. Both systems had their merits and had been shown to be suitable for the intended application, so our decision was based on how the membrane could be best integrated into the system as well as the supplier’s ability to meet overall project timelines for development.
One additional feature we added to our ultrafiltration assembly was a closed permeate-collection system. We had deemed it impossible to integrity test the microfiltration membrane with the same rigor as is possible with a sterilizing-grade filter. Thus it could not be regarded as a sterile barrier. Keeping the permeate within the sterile envelope eliminated the need for integrity testing of that membrane.
System Sterilization: An additional consideration to the equipment design is sterilization of custom-designed disposable units. Most disposable items are sterilized by gamma irradiation, which like all sterilization processes needs to be validated. Validation may be achieved by monitoring dose levels in loads and then releasing equipment as having received dose levels >25 Kgray. Or these studies can be supported by the inclusion of biological indicators. That approach can be more expensive and time consuming, but in our experience it allows a vendor to release equipment with a certificate of sterility rather than as merely having received appropriate dose levels. In all cases, the materials and dose levels used have to be such that component integrity and performance are not compromised.
Equipment OptionsHaving arrived at an outline design for this manufacturing system, we discussed it with potential manufacturers, who proposed two design strategies: One was based predominantly on preexisting units that would be connected using tube welders, and the second was a modular approach in which units would be connected by sterile connector units wherever feasible to reduce the requirement for tube welding.
A key driver for this project, as with all development programs, was the time it would take to design, procure, and validate the systems. This was as important as the cost and achieving an optimally designed system. In fact, it was predominantly this driver that the headed our selection of vendor. The decision was made to use Spectrum membranes supplied by JM Separations (www.jmseparations.com) based on larger assemblies rather than the modular approach proposed by GE Healthcare (www.gehealthcare.com). So this is no reflection of the suitability of the GE products for a program of this type.
ValidationMuch of our initial design had been performed as a paper exercise, with review of the outline drawings, also based on some preexisting modules that had been assessed as part of the initial development studies. No fullscale development units were actually generated at this stage. Once the outline design had been agreed upon, it was necessary to build a validation program around the development and approval of those outline designs and then direct the next stages of the program for transferring the process from a development into a manufacturing program. Validation of the units was structured into three phases: equipment qualification, a process simulation using tryptone soya broth (TSB), and shipping qualification. The latter was included because the product was being filled at a third-party site in the United States, and it was necessary as part of the sterile chain for product manufacture, but it will not be discussed here.
The predominant part of our validation efforts related to the equipment qualification and TSB simulation (Table 1). For a disposable system, qualification relies heavily on vendor assurance and documentation regarding materials of construction and sterility. Because this product was being used for a phase 2 clinical study, we deemed the requirement for in-depth studies on process leachables and extractables to be unnecessary.
Table 1: Outline of validation plan

Our in-house validation studies relied heavily on the performance of TSB simulation. This approach allowed for a combination of PQ and broth-fill validation to be performed as a combined operation. With an early stage clinical product, the aim of this validation study was to verify monosepsis and product safety rather than validate the manufacturing process itself.
Before those PQ studies, two critical activities need to be performed. First, process-related standard operating procedures (SOPs) and batch manufacturing records (BMRs) needed to be written. Because these documents were being generated for a de-novo process, it was accepted that they would require revision, which was handled under standard change-control systems.
The second activity was training of the manufacturing staff. This is widely recognized as a critical requirement when introducing disposable systems into manufacturing operations. With a novel process such as this one, the training was approached at three levels: to familiarize the operators with the process itself, then to train staff on the disposable assemblies being used for the manufacturing operation, and finally to train staff in individual process operations, with specific reference to critical operations such as tube welding.
Our validation studies were completed successfully and showed that the process could be performed both aseptically and reproducibly. This implied that it should be possible to retain a monoseptic process using this disposable approach to the manufacture of live-bacterial products. On completion of the validation, we initiated a manufacturing campaign based around the performance of a full-scale engineering run followed by the manufacture of the clinical batch. The engineering run was included because we felt that it would be essential to assess the process at scale and thus allow for any required revisions of the process and related documents. So we compared the results obtained (Table 2) to the process development works. Our conclusion was that the process was scalable and that it had generated highly comparable results.
Table 2: Yields from process development, engineering, and GMP manufacturing runs

The first and most important conclusion to be drawn from this project was that it is possible to develop and implement custom disposable manufacturing for production of a monoseptic product. Our results met not only the customer’s requirements, but most important, it met regulatory requirements for products of this type. It also provided a platform for scalable manufacturing that could be used for late-stage and in-market supplies and was regarded both by the customer and project team as a significant achievement and a successful conclusion to the contract.
In reflecting on a project like this, however, a number of lessons can be learned. Regarding the initial feasibility studies performed ahead of process assembly design, it is clear that more studies would have been useful to fully design operational windows and parameters so to identify the impact of process times and conditions on scale-up. As with all process transfers, operational timing inevitably increases, and the impact of such needs to be assessed.
The most important lessons can be learned from our process and approach for designing the equipment. The first of these is to appreciate the full complexity of the challenge and the time and effort required to produce a final system design and then implement it into a manufacturing operation. Design of conventional stainless-steel assemblies is time consuming, involving not only the full assessment of piping and instrument designs (P&IDs), but also a general assembly (GA) drawing before equipment fabrication, followed by a tack-welded preassembly at the fabricator in advance of final construction. This process allows performance of process flows and hazard assessments as well as a three-dimensional layout of the equipment to be determined before construction. Although such a process is not perfect, it does provide opportunities for operator input into the design. Assessment of operating requirements can be made and design changes completed. One major drawback with custom disposable equipment is the temptation to see them as simple systems and thus skipping or significantly abbreviating a number of the conventional design steps.
From an operational point of view, the major failing of the system we used was in our incomplete appreciation of the challenges it would present to its operators. Specifically, we should have foreseen what the system would look like (and how large it would be) when assembled, as well as the issues those operators would encounter when handling bags and performing critical activities such as tube welding. We needed to be able to generate mock-up systems with the component parts and perform basic development runs before agreeing on the final design.
This may appear obvious, but disposable systems do not always allow such an approach because of the high costs of manufacturing single units and the time required to produce custom products — as well as the vendor’s capacity (and in some cases willingness) to generate these alongside standard product ranges. If this had been done, then it is likely that we would have selected a more modular system in which component parts could be attached and removed as required and preformed sterile connectors used where feasible. Our operators found the preassembled units to be unwieldy and difficult to manipulate, which created a difficult operating environment that could have led to an increased level of processing errors. This is not the supplier’s fault, but rather it relates to specifications we should have prepared. It also highlights the need for integrating technology transfer and production staff into all phases of a project as early as possible.
A final lesson learned was in regard to the overall time requirements of design, manufacturing, and validation studies. Gamma-radiation sterilization processes can be complex if they are not performed by a vendor. Even this apparently simple system took nine months to design, fabricate, and validate.