Single-use bioprocess manufacturing systems increasingly are being implemented by the biopharmaceutical industry based on safety, time, and cost-reduction benefits. These disposable systems are used to process or contain fluids ranging from culture media, additives, and buffers, to bulk intermediates and final formulations. In many cases microbial control or sterility is required to ensure product purity and safety. Radiation sterilization is a common means of microbial control and sterilization applied to single-use systems. The standard methods for validating radiation sterilization are not widely understood in the pharmaceutical industry, which has historically relied on moist heat (steam) for sterilization of stainless steel bioprocess systems.
Under the auspices of The Society of the Plastics Industry, Inc. (SPI), the BPSA Technology Committee established a subcommittee to develop this guide to irradiation and sterilization of single-use bioprocess components and systems.
Scope, Purpose, and BackgroundScope: This guide is intended to provide basic information and general recommendations on gamma irradiation for microbial control and sterilization of single-use disposable bioprocess systems applied to biopharmaceutical manufacturing. Although e-beam irradiation is similar to gamma irradiation and follows the same standards, it is generally not used for microbial control or sterilization of single-use systems because of its limited penetration ability and thus is not considered in this document.
Purpose: The intention of the BPSA Irradiation and Sterilization Subcommittee is that this guide will
-
Educate readers on the basics of gamma irradiation and two approaches for reducing or eliminating bioburden on single-use disposable bioprocess systems: microbial control and sterilization validation.
-
Enable readers to understand the differences between microbial control and validated sterilization, to differentiate where each is applicable to single-use biopharmaceutical manufacturing, and to make educated decisions about which may be sufficient or required in different biopharmaceutical manufacturing process applications.
-
Summarize the standard methods for validation of sterilization of health care products by gamma irradiation, which are recognized as applicable to single-use systems for the biopharmaceutical industry.
-
Provide recommendations on bioburden and sterility test methods used in validating radiation sterilization of bioprocess components and systems.
Background: Industry standards for validation of sterilization of health care products by irradiation have been established by organizations such as the American National Standards Institute (ANSI), the Association for the Advancement of Medical Instrumentation (AAMI), the International Organization for Standardization (ISO), and ASTM International (formerly the American Society for Testing and Materials). Advancements in the biopharmaceutical and biotechnology drug industry have led to the need for presterilized or microbially controlled products that can be directly incorporated in critical manufacturing processes. Initially, these disposable products were targeted for small-scale applications such as laboratory-scale drug development and preclinical studies.

DONALD GRUENER (WWW.ISTOCKPHOTO.COM)
In recent years, an exponential growth has occurred in clinical and process-scale single-use disposable product lines, including high-area filter capsules, membrane chromatography units, tubing, connectors, and flexible biocontainers (e.g., polymeric film bags). The established industry standards for validation of irradiation sterilization of health care products have been applied successfully to such products when a sterile claim is desired and are recognized by device and drug regulatory agencies.
Application of sterilization validation standards for health care products to bioprocess systems, however, can be quite costly and complex. As discussed further in this guide, use of model master systems, supported by the standards, can ease these burdens, or irradiation without a sterility claim may be used in cases where a high but undetermined degree of microbial control is sufficient. Key definitions are highlighted in the “Glossary” box.
Basics of Gamma IrradiationGamma irradiation is the application of electromagnetic radiation (gamma rays) emitted from radionuclides such as Cobalt 60 (60 Co) and Cesium 137 (137 Cs) isotopes. Gamma rays are not retarded by most materials and can penetrate through most single-use bioprocess system components. Microorganisms are inactivated by damage to their nucleic acids resulting from this ionizing irradiation. Gamma rays are also not retained by material and leave no residual radioactivity.
Gamma irradiation dosage is measured in kilogray (kGy) units, which quantify the absorbed energy of radiation. One gray is the absorption of one joule of radiation energy by one kilogram of matter (one kGy = one joule/gram). Dosages ≥8 kGy are generally adequate to eliminate low bioburden levels (Table 9 in Reference 2). In cases where bioburden level is elevated (>1,000 colony forming units, or cfu, per unit), as may occur with very large single-use systems, higher doses may be required to achieve sterility.
Generally, 25 kGy can achieve sterility with a sterility assurance level (SAL) of 10−6. Even with elevated bioburden levels, bioburden reduction can be achieved with lower probabilities of sterility (e.g., SAL of 10−5 or 10−4). Products irradiated to such SALs are still sterile but have higher probabilities of nonsterility and may not meet standards for validated sterile claims as specified in industry standards for sterilization of health care products.
The gamma irradiation process uses well-defined operating parameters to ensure accurate dosing. In a well-designed irradiation facility, for any given density of material the only variable determining the amount of radiation the product and microorganism receives is the time the material spends within the radiation field. Products are not exposed to heat, humidity, pressure, or vacuum. Gamma irradiation produces minimal waste byproducts and does not require quarantine for outgassing (as with ethylene oxide gas sterilization) or routine biological reactivity testing. As a constant and predictable sterilization method, gamma irradiation provides benefits in safety, time, and cost.
In addition to microbial inactivation, gamma irradiation also causes ionization and excitation of polymer molecules (7). Over time and dependent on dose, the adsorbed dose can result in changes to the physical or chemical properties of the polymers. Although some polymers show higher resistance to irradiation-induced changes than others, all polymers are affected to some degree. Whereas a minimum dose of 25 kGy is desired for microbial control or sterilization, the actual applied dose is often in the 25–50 kGy range. It is important to note that adsorption of radiation is cumulative and that some polymers that may be qualified for use in gamma-irradiated single-use bioprocess systems may not be capable of withstanding more than 50 kGy and still retain their integrity over time. Consequently, repeated irradiation of single-use systems or components should be avoided (8).
Microbial Control and SterilizationThe general concepts and protocols described in the current industry standards for sterilization o
f health care products by gamma irradiation can be directly applied to single-use bioprocess systems. These industry standards require validation of the efficacy and reproducibility of the sterilization process, based on determination of average bioburden and subsequent sterility testing of systems after minimal radiation dose exposures. Systems validated as sterile are also subject to routine audits involving bioburden and sterility testing. Standard procedures for gamma sterilization validation and application to single-use bioprocess systems are summarized in Sections 6 and 7, respectively.
Application of these standards to bioprocess systems manufactured in small custom batches, especially for prototype systems used during preclinical or clinical drug process development, can be costly and burdensome and may require revalidation of sterilization of the final production design. As an alternative to a sterile label claim, many bioprocess systems may be adequately microbially controlled simply by irradiation typically in the range of 25 kGy based on dosimetry, and the standard methods for sterilization validation may not be required.
The minimum 25 kGy sterilizing dose claim originated from a study performed by Charles Artandi and Walton Van Winkle in 1959. The authors determined the “minimum killing dose” for over 150 different species of microorganisms. As a conclusion of their study, they chose 25 kGy as the sterilizing dose, stating that “this [25 kGy] is 40% above the minimum to kill the most resistant microorganisms” (9). Consequently, 25 kGy became established as a suitable minimum irradiation dose for sterilization. This historical sterilizing dose of 25 kGy also can be sufficient to eliminate viable bioburden and provide a high level of microbial control when a validated sterile claim is not required.
Components or systems requiring zero or low bioburden when applied in nonsterile processes do not require a validated sterile claim and may be qualified as microbially controlled. The following section is intended to help readers determine whether validated sterilization, or more simply, microbial control, is appropriate.
Bioburden Control of Single-Use Systems in ManufacturingBiopharmaceutical manufacturing, in which drug products are produced by cell culture, can be divided into four processing stages (Figure 1). In the upstream stage, nutrients and other fluids, along with the producer cells, are introduced to a fermentor or bioreactor, and the cells produce the target molecule. In the harvest stage, cells are separated from the target molecule using centrifugation, depth filtration, and/or membrane filtration. In the downstream stage, the target molecule is subjected to a series of separation, purification, and concentration stages applying chromatography and membrane filtration to ultimately produce a purified bulk drug product. In the final formulation and fill stage, the purified bulk (active pharmaceutical ingredient (API) or biological) is prepared in a final liquid formulation, sterilized by filtration, and/or aseptically filled into final sterile containers. This latter stage is similar to how synthetic pharmaceuticals are manufactured by aseptic processing.
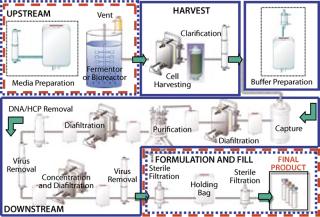
During the course of these stages, there is an ongoing need for microbial control to prevent adverse contamination. International regulations state that processes claimed as sterile must be validated to support the sterilization claim. This does not apply, however, to processes claimed as microbially controlled, which also may have zero or low bioburden but have not been validated with a defined SAL. Although the high assurance of sterility provided by validation is required of bulk drug claimed as sterile and of final sterile formulation and filling of injectable drugs and biologics, many preparative stages in bioprocess manufacturing (Figure 1) are difficult to sterilize and may not require actual validated sterility. Instead, they are operated under a high degree of microbial control, as can be provided by gamma irradiation without sterilization validation.
Consideration of whether a process step must be validated as sterile, in contrast with maintaining a microbially controlled (zero or low bioburden) state, is critical to optimizing the time and cost requirements for process development. Similarly, when validated sterility is required, consideration whether an entire single-use system or only the fluid path needs to be validated as sterile also can affect time and cost requirements. As mentioned previously, establishing and maintaining a validated claim of sterility for single-use bioprocess systems or components can require significant cost that will be ultimately borne by their users. Particularly during process development and often during subsequent production, a claim of microbially control based on irradiation may be sufficient to prevent significant microbial contamination while also saving the time and cost of establishing and maintaining a validated sterile claim.
This balance between added assurance of sterility afforded by a validated sterile claim and the substantial added cost to attain that added assurance is one that must be made by biopharmaceutical manufacturers. The choice must be made case-by-case and is most likely to be driven by a risk assessment of each unit operation as well as by the manufacturing process as a whole.
In upstream processing, bacterial (e.g., Escherichia coli) cell cultures tend to be operated in short time frames and are fairly resistant to overgrowth by low levels of contaminant bacteria. In such cases, a user may consider that single-use disposable bioreactors and culture media or other nutrient additives prepared in single-use systems may be safely operated under microbial control provided by irradiation without sterilization validation. Mammalian cell cultures, by contrast, are often run for extended periods of time (e.g., up to two weeks) and are generally more sensitive to bacterial contamination. In such cases, the additional assurance provided by sterilization validation may be merited, although the high but unspecified probability of nonsterility afforded by gamma irradiation for microbial control may be adequate during process development and even production.
Cell harvesting and downstream processing steps (centrifugation, depth or membrane filtration, and chromatography, with intermediate holding) are rarely validated for sterility due to complexities and/or limitations of their equipment, especially during process development. Instead, process equipment is chemically disinfected or sanitized and maintained as microbially controlled for zero or low bioburden. Intermediate holds can be kept at low temperature to prevent microbial growth before a further nonsterile process step. In cases where these process steps are not claimed to be sterile, it is unnecessary to validate sterility of single-use systems for preparation of process buffer feed solutions or intermediate holds. Buffer solutions or intermediates filtered through irradiated bioburden reduction filter systems as microbially controlled feeds are generally suitable for process steps not validated as sterile.
Formulation and fill also provide options for choosing b
etween validation of sterility and microbial control by irradiation. When a nonsterile finished bulk active pharmaceutical ingredient or biological is stored under reduced temperature before subsequent processing including formulation, sterilizing filtration, and final sterile filling, prefiltration storage in microbially controlled containers may be adequate. The only key steps clearly requiring single-use systems to be validated as sterile are in the preparation of sterile API and the aseptic filling of sterile containers; that is, if the fluid is to be claimed as sterile, then the single-use system it is filled into must have that validated claim.
Several industry standards are used for sterilization validation of gamma irradiated health-care products. The ANSI/AAMI/ISO 11137:2006 standard (Sterilization of Health Care Products — Radiation) was published originally in 1994. It was followed by several supporting technical reports developed by ISO and AAMI. Key among those is AAMI Technical Information Report (TIR)33:2005 on Substantiation of a Selected Sterilization Dose — Method VDmax (10).
During systematic review of ISO 11137:1994 (adopted in the United States as ANSI/AAMI/ISO 11137: 1994), the document was revised and divided into three parts under the general title, Sterilization of Health Care Products — Radiation. The three parts of ANSI/AAMI/ISO 11137:2006 are described below.
Part 1: Requirements for Development, Validation, and Routine Control of a Sterilization Process for Medical Devices specifies requirements for development, validation, process control, and routine monitoring in the radiation sterilization for health-care products. Part 1 applies to continuous and batch-type gamma irradiators using the radionucleotides 60 Co or 137 Cs, and to irradiators using a beam from an electron or X-ray generator.
Part 2: Establishing the Sterilization Dose describes methods that can be used to determine the minimum dose necessary to achieve the specified requirement for sterility, including methods to substantiate 15 or 25 kGy as the sterilization dose.
Part 3: Guidance on Dosimetric Aspects provides guidance on dosimetry for radiation sterilization of health-care products and dosimetric aspects of establishing the maximum dose (product qualification); establishing the sterilization dose; installation qualification; operational qualification; and performance qualification.
ANSI/AAMI/ISO standards describe two methodologies for sterilization validation:
-
Dose setting methods (Method 1 or Method 2, ANSI/AAMI/ISO 11137), which take into account distribution and radiation resistance of product bioburden, and to a limited extent, the end-use of the product, and
-
Dose substantiation methods, (VDmax methods), which entail experimentation designed to qualify predetermined gamma dosages as a sterilization dose (1,2,3).
-
Some of those methods’ similarities include
-
Application only in conjunction with a suitable quality system
-
Recognition that dose control and bioburden control are two equally important aspects to sterility assurance
-
Requirement for demonstration of continued effectiveness through periodic testing to verify that the established irradiation sterilization dose remains appropriate for product sterilization.
Table 1 summarizes additional similarities and differences of the various methods. Although it illustrates the main attributes of different methods, please refer to the actual ANSI/AAMI/ISO standards or AAMI TIR for comprehensive information.
Table 1: Comparing standard irradiation sterilization methods for healthcare products
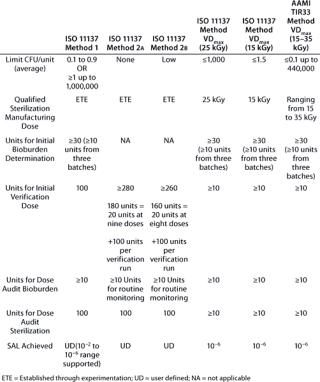
Collectively, these methods provide users with a degree of flexibility in application of standards to qualify an irradiation process, in recognition that products are not all manufactured and commercialized under uniform circumstances. Users are afforded latitude to make pragmatic decisions, provided that they observe technical correctness in several key areas:
-
Flexibility to qualify sterilization of elements such as individually packaged units, kits, and components
-
Flexibility to qualify sterilization of the entire unit or of a part of a unit (e.g., fluid path only)
-
Use of product sample size required for irradiation cycle qualification.
-
Acceptable bioburden levels presterilization
-
Frequency of audit testing (performed periodically to support continued use of the established sterilization dose).
Although AAMI TIR33:2005 and ANSI/AAMI/ISO 11137 standards were initially developed with a focus on the medical device industry, they also apply to bioprocess systems and components. The remainder of this guide is therefore founded on these industry standards.
GLOSSARY
Aseptic: Technically, free from disease-producing microorganisms. In bioprocess applications, it refers to an operation performed in a controlled environment or with purpose-designed connectors to prevent contamination through introduction of microorganisms (may be sterile or microbially controlled).
Bioburden: Population of viable microorganisms on a product. In the context of sterilization, it should be determined immediately before sterilization. Bioburden may be described for an item in its entirety, including external surfaces, or for fluid contact surfaces only.
Bioprocess Components: Single-use disposable parts, modules, or sections of a system, which may include but not be limited to filters, membrane chromatography units, tubing, connectors, fittings, flexible bags or rigid containers, and probes.
Bioprocess System: An assemblage or combination of bioprocess components intended to facilitate the processing of pharmaceutical or biological solutions in a single-use disposable format. The system may be preassembled before a bioburden reduction/microbial control step or assembled by a user by connecting presterilized subassemblies through the use of aseptic connections.
Dosimetric Release: The determination that a product is sterile based on physical irradiation process data rather than sterility testing.
False-Positive Sterility Test Result: Test result that is incorrectly interpreted to indicate that the test article contained viable organisms and that the article was not sterile. Commonly, such a result will derive from faulty sample handling or test execution — or from an interaction between a sample and the microbial growth medium that causes the medium to become turbid.
Irradiation: Application of ionizing radiation capable of destroying microbial bioburden.
Microbial Control: Assembly of products in a controlled clean environment followed by exposure to gamma radiation. This process reduces bioburden load but does not support a “steri
le” label claim.
Sample Item Portion (SIP): Defined portion of a product assembly or component.
Sterile: Free from viable microorganisms.
Sterility Assurance Level (SAL): In practice, the sterile state cannot be proven. Instead, sterility is expressed as the probability of a single viable microorganism occurring on an item after sterilization (1). Normally the term is expressed as 10−n and not as an absolute.
Sterility Testing: Tests performed to determine whether viable microorganisms are present. Commonly, the test involves immersing a component or system or flushing a fluid pathway with sterile microbial growth medium, incubation of the medium under conditions favorable for microbial growth, and observation of turbidity or other indication of microbial growth after a suitable incubation period.
Sterilization: A validated process used to render a product free from viable microorganisms. It is generally accepted that a terminally sterilized unit purporting to be sterile attain a sterility assurance level of ≤ 10−6; that is, probability of less than or equal to one chance in one million that a viable microorganism is present in the sterilized unit (1,2,3,4,5,6). Lower SALs may be validated as sterile in some cases (2).
Validation: Establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its predetermined specifications and quality attributes.
The dose-setting methods entail experimentation that leads a manufacturer of the sterile product to a qualified minimum irradiation dose. The general approach when using this type of method is described in ANSI/ AAMI/ISO 11137:2006 and comprises the following steps:
-
Determine the target SAL.
-
Obtain a specified number of products for testing from three production lots (Table 1).
-
Experimentally determine average bioburden following ANSI/AAMI/ISO 11737-1 (11, 12).
-
Determine the verification dose appropriate for the bioburden.
-
Perform a dose verification study (irradiation and sterility testing).
-
Interpret results — accepting or rejecting the study.
-
Establish the minimum dose for sterilization, based on the bioburden and SAL requirements.
-
Institute periodic dose audit testing for continued effectiveness.
Dose-setting methodologies involve qualification of the dose based on the actual radiation resistance of microorganism isolated from the product. As a result, these methods provide the lowest possible dose but require more product samples than a dose substantiation method.
Dose Substantiation MethodsThe ANSI/AAMI/ISO 11137–2:2006 VDmax methodology allows for substantiation of two preestablished irradiation sterilization dosages: 15 kGy and 25 kGy. AAMI TIR33:2005 provides the flexibility of using seven additional sterilization doses: 17.5, 20, 22.5, 27.5, 30, 32.5, and 35 kGy. The sterilization doses range from 15 kGy for bioburden up to 1.5 cfu, 25 kGy for bioburden up to 1,000 cfu, and 35 kGy for a bioburden up to 440,000 cfu. A manufacturer should maintain strict bioburden controls at every stage of its manufacturing process. Other than the additional dose set points, ANSI/AAMI/ISO 11137–2:2006 and AAMI TIR33:2005, are based on identical principles and provide essentially identical guidance.
The general approach when using the VDmax method comprises the following steps:
-
Obtain products for testing from three production lots.
-
Experimentally determine average bioburden. The bioburden estimate must meet the criteria for the selected VDmax dose.
-
Determine the verification irradiation dose appropriate for the average bioburden.
-
Perform a dose verification study (irradiation and sterility testing)
-
Interpret results — accept or reject the study.
-
Accept or reject the predetermined irradiation dose as the minimum dose for sterilization.
-
Institute periodic dose audit testing for continued effectiveness.
The VDmax methods substantiate an irradiation sterilization dosage to achieve an SAL of 10−6; higher SALs (lesser assurance of sterility) are not supported currently by the method. The fixed dosages are keyed off of two inputs: bioburden number (determined experimentally) and a data-driven assumption of worst-case bioburden resistance (2). Although the choices of sterilization dose in AAMI TIR33:2005 are fixed, the sampling requirement is smaller than Method 1 and 2 of ANSI/AAMI/ISO 11137.
Approaches to ValidationSingle-use bioprocess systems and components pose a challenge during validation testing due to their size and complexity. A large-scale system could be, for example, an assembly of a 10,000-L bag connected to several meters of tubing, high-area filter capsule(s) that could be 254 mm (10 inches) or larger, and a connector with a complex design — or perhaps a complex manifold of connectors. Thus, you should carefully plan an easy and scientifically valid approach to bioburden and sterility testing. The sample item portion (SIP), product groupings, and family approaches to testing described in the ANSI/AAMI/ISO 11137:2006 guidelines offer strategies addressing the complexity of medical-device designs and health-care products, and are also applicable to large-scale bioprocess components and systems as outlined in the sections described below.
Product Groupings/FamiliesThe simplest strategy is for three standard manufacturing lots to be sampled. These lots should not deviate from standard manufacturing conditions. Commonly, manufacturers of multiple products develop one or more product families with the intent that a single product can represent the remaining members of its family (10). This single product used to represent a product family may be designated as a “master product,” an “equivalent product,” or a “simulated product.”
Master Product: A master product is not necessarily the largest in a family. In cases where all materials and components are the same and products vary only in size or amount of materials, the master product is usually the largest one. Depending on the materials and manufacturing processes included, a master product may be one that possesses the most component types, the greatest combination of materials, the highest filter area, the most handling during manufacture and assembly, or a combination of those factors. Ultimately, the master product is the one deemed most challenging to a sterilization process.
Equivalent Product: Equivalent products are not necessarily those that possess identical bioburden levels, because bioburden levels naturally vary between samples and sets of samples taken over time. Products can be considered equivalent if
-
Bioburden estimates are about the same
-
Bioburden characterization is similar, and
-
Bioburden estimate results in a sterilization dose that is about the same when using Method 1 and Method 2, or a verification dose that is about the same when using VDmax.
Simulated Product: A simulated product is one that contains the same materials and components of an actual product but has been fabricated for test purposes only and thus is not an actual product. This is typically fabricated to inclu
de or combine representative worst-case attributes of a product family. One example is an assembly made with an excess of tube runs to account for different tube configurations represented in aggregate by the product family. Rejected or discarded material can be used to fabricate simulated products as long as the bioburden of the resulting simulation is representative of actual products, and the composition or configuration of the simulated product does not affect the validity of the dose determination. In designing a simulated product, care should be taken to create one that does not create an undue challenge to the laboratory personnel who perform bioburden and sterility testing.
Bioburden and sterility testing of bioprocess systems or components present two specific challenges. First, large articles are especially difficult to manipulate aseptically. Specific issues include introducing such large articles into a biological safety cabinet, aseptically adding and removing fluids to the test article, aseptically handling large fluid volumes required for testing, and providing incubation space for large systems. For testing the entire product (rather than the fluid path alone), the additional issue of handling its external surfaces without contaminating them must be overcome. The second issue is that bioprocess systems or components tend to be quite expensive. Taken with the likelihood that such products are sold at very low volumes in the beginning of their life cycle, testing a relatively large number of actual articles could be prohibitive to their commercialization.
Particular approaches that manufacturers take to address these issues must be developed specific to each case because they are typically a balance between
-
A desire to ensure technical correctness by testing the actual article, and
-
A desire to avoid false results (inflated bioburden estimates or false-positive sterility test results) caused by handling of unwieldy articles.
Several common approaches can lessen the difficulties posed by those two issues. The following examples are intended to illustrate what may be done to overcome the issues; they are not intended to exclude other scientifically defensible approaches.
Sample Item Portion (SIP): The SIP approach allows for manufacturing and testing a reduced-scale product. This reduces both the cost of the test article and difficulties with manipulating product in the laboratory. This approach increases experimentally determined bioburden estimates by a multiplier based on scale differences between the SIP article and the actual product represented. SIP multipliers can be determined through experimentation or simply by taking a ratio of the model to the largest system manufactured.
Fluid Path: In some cases, it is appropriate to validate the sterility of a product’s fluid path. Testing the fluid path may be simpler than testing an entire product because a product acts as its own barrier to contamination. A laboratory operator has greater freedom to manipulate such products without concern for contamination.
Bioburden testing of a fluid path generally entails partially filling the product with a sterile buffer to ensure that all surfaces are wetted, then agitating the article by hand to promote suspension of organisms into the buffer. The buffer then is removed and tested for bioburden load through standard microbiological methods. As a means of better quantifying bioburden, a bioburden recovery study is performed before the actual bioburden study is initiated. The recovery study serves two purposes: quantifying the efficiency with which bioburden is recovered from the test article and helping to define the specific methods that will be used in the actual bioburden study.
For sterility testing, the article is either partially filled with microbiological growth medium or with an extraction fluid. Following an agitation step, the growth medium can be left inside or removed and assayed for microbiological load.
Sectioning of a Large Product: To aid in handling, a large article can be split into parts that are more easily manipulated in the laboratory before testing or irradiation. This approach is especially effective if an entire product and not simply a fluid path is to be tested. One example of this approach is cutting a large biocontainer into 10 sections, dicing each into several small pieces and adding the pieces to a container that is easy to handle aseptically in a microbiology laboratory and biological safety cabinet. In this situation, each container would be tested, and the sum of the bioburden would represent the bioburden of the original bag. Similarly, all 10 containers would need to be sterile for the original biocontainer to be counted as sterile.
Use of Product Packaging As a Containment Device: Bioprocess systems or components generally are packaged in two outer bags to ensure their cleanliness and sterility. Some articles, especially those for which the entire product must be tested, are well suited to testing while they remain in their original packaging. In this scenario, the inner packaging serves as the containment vessel. A small opening is made in the packaging to access the test article and to add or remove buffer or growth medium. The opening is then aseptically sealed to maintain a closed system and prevent microbial contamination. This method is less effective for test articles that are closed-loop systems and must be opened to access internal surfaces.
Dosimetric ReleaseIn qualifying a gamma irradiation microbial control or sterilization cycle, you must have the means to quantify the gamma irradiation dose. This is done through dosimetry, a field relating to the quantification of absorbed radiation. The measurement devices are referred to as dosimeters. Dosimetry has specific importance in two areas of qualification:
-
The verification dose used to verify a specific sterilization dosage must be measured accurately (to ± 10%)
-
The load should be dose-mapped to ensure that uniformity of gamma dosage is sufficient for the configuration undergoing irradiation.
Although the science behind dosimetry is beyond the scope of this guide, various dosimetry technologies exist. Users should ensure that the dosimeter and operator are qualified to provide sufficient accuracy and precision, especially in the range of radiation dose under consideration. This method validation concept is applied as it would be to any measurement method yielding data on which product qualification and release may be based. Typically, specialized service providers of gamma irradiation perform dosimeter qualification.
After an irradiation cycle is validated, it may be used in production of microbially controlled or sterile products. Product is released after gamma irradiation on the basis of dosimetry: a dosimetric verification that the cycle yielded a required minimum dose to the product load. No routine sterility testing is required for lot release. Because product is released dosimetrically, its manufacturer must ensure that the dosimetry practiced by the gamma irradiation service provider is well qualified and that the gamma irradiation operation is contr
olled to consistently deliver the correct dose range.
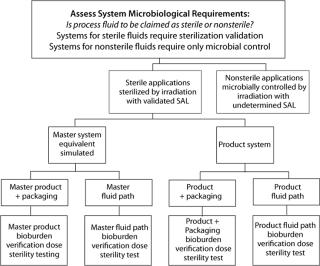
Disposable bioprocess systems represent a substantial benefit to biopharmaceutical process development and manufacturing. Accompanying the benefits are new decisions for all stakeholders in the adoption and incorporation of these systems. With respect to quality attributes such as microbial control and sterilization, some issues may be addressed in general terms, whereas others should be addressed in the context of a specific biopharmaceutical manufacturing process. Figure 1 provides a decision tree for sterilization and microbial control.
This guide presents a discussion of the fundamental issues related to gamma irradiation and the level of microbiological control practiced in the manufacture of disposable bioprocess systems and components. It is intended to frame the microbiological control issues and to provide a structure for making informed and rational decisions as to the level of microbiological control required in the manufacture and use of disposable bioprocess systems.