Significant changes are sweeping the vaccine manufacturing industry. Demand for human vaccines is predicted to grow significantly — in part driven by needs in emerging countries, where only small fractions of their large and growing populations has access to vaccines. Sustained growth is expected to yield a vaccine market of US$25 billion by the year 2015 (1).
Relatively low immunization rates in the Asia–Pacific regions represent significant untapped potential for vaccine manufacturers. Growing populations, increased government funding, and increasing personal wealth leads to more money being spent on improving personal health overall. Estimates of vaccine growth range as high as 65% in the Asia–Pacific. Significant vaccine-related investments are flowing into the region as many industry leaders establish research and development centers and expand their Asia–Pacific manufacturing capacity. China’s National Development and Reform commission is partially sponsoring investments and notes that development of new vaccines is the “number one project” in the biopharmaceutical industry.
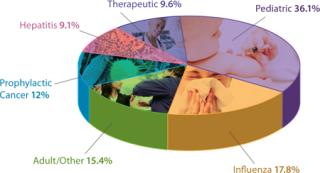
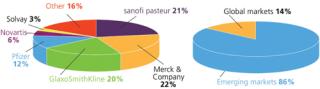
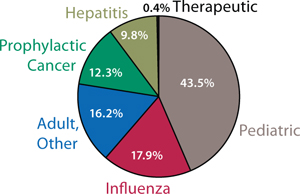
In addition to demand from emerging markets, major drivers of growth include global unmet medical needs and growing interest in therapeutic vaccines (Figure 1). Both established manufacturers and newcomers are investing significant resources into vaccine development; a recent study tallied 200–250 companies developing more than 600 vaccine products (1). Vaccine manufacturing is dominated by several key players including Sanofi Pasteur, Pfizer, GlaxoSmithKline, and Merck (Figure 2, LEFT). Most manufacturing is located in emerging markets (Figure 2, RIGHT).
The Need for New Approaches
Increasing and evolving demands for all types of products creates new challenges for vaccine manufacturers. As the market grows, innovative approaches to development and production will be needed to accelerate delivery of novel products.
The pressure to indentify new approaches is perhaps most intensely felt by manufacturers of influenza vaccines and others that may be required on short notice, in large quantities, and in response to public health emergencies. The 2009 H1N1 influenza pandemic made clear the need for more rapid development, increased responsiveness, and a shift of production closer to population centers.
Health policy-makers are encouraging the use of new technology to meet these challenges and make vaccines more accessible. Improvements to influenza vaccine manufacturing processes will certainly be adopted by companies making other types of vaccines, so these efforts ultimately will have widespread value.
In August 2010, the US President’s Council of Advisors on Science and Technology (PCAST) presented a report on reengineering the influenza vaccine production enterprise (2). That report addressed concerns related to the 2009 H1N1 influenza. A quantity of doses sufficient for half the population became available at 38 weeks, and supply adequate to protect the entire US population would have taken ~48 weeks to produce. The council considered that timeline to be too slow by three to five months. So the council recommended a number of steps to reduce the time needed to deliver pandemic flu vaccines by streamlining methodologies for producing seed virus; making needed reagents for potency tests; testing the sterility of vaccine preparations; and placing vaccine materials in appropriate delivery devices. Accelerated Sterility Testing
The PCAST report included the following statement regarding sterility testing of initial H1N1 vaccine lots: “Initial lots of H1N1 vaccine were placed in syringes and vials or nasal sprayers and packaged by September 30. Given the urgency associated with the developing pandemic, manufacturers shipped 2009 H1N1 vaccines before sterility testing was completed, recognizing that some lots might need to be recalled.”
The council highlighted a need to shorten the time required for sterility testing of vaccines. It directed the US Food and Drug Administration (FDA) and the Biomedical Advanced Research and Development Authority (BARDA) to fund applied research that will adapt rapid nucleic acid amplification and sequencing methods to test for sterility. This would reduce or eliminate manufacturers’ dependence on classical microbial assays.
Each batch of vaccine material must pass a sterility test to ensure that it is not contaminated with bacteria or fungi. Although current methods of doing this are reliable, they can take several weeks to complete. Faster sterility test methods are available — based on array-based sequencing methods and other technologies — that could shorten the time before vaccine release. Although research and development of such methods for microbial contamination continues, rapid methods for mycoplasma are already available.
Mycoplasma bacteria infect eukaryotic cells and embryonated chicken eggs, disrupting their growth and metabolism. The presence of mycoplasma in vaccine production processes can cause decreased protein quality and yields. If undetected, it can cause adverse side effects in patients. Detecting mycoplasma from biopharmaceutical manufacturing processes is extremely difficult because such contaminations do not generally cause pH changes or visual turbidity in media. Removal is equally challenging because of the small size and pleomorphic nature of mycoplasma.
Traditional growth-based methods that test for mycoplasma contamination take as long as 28 days, which poses a problem for vaccine manufacturers. If multiple batches have been pooled already and a contamination event is identified, the entire lot would need to be scrapped. Time-consuming methods do not allow for appropriate investigations, which compounds the problem by making root-cause analysis for a contamination very difficult and time-consuming.
The compendial mycoplasma test method involves both culture and indicator cell tests. The culture test takes place over the course of 28–35 days, whereas the incubator test requires seven to 10 days. Newer methods to detect mycoplasma leverage nucleic acid techniques (NATs). Most such methods are based on amplification of a mycoplasma’s nucleic-acid content using enzymes. Currently available methods include polymerase chain reaction (PCR), real-time transcription mediated amplification (real-time TMA from Gen-Probe Incorporated), and microarray-based detection methods.
For a method that requires multiple species to be detected, ribosomal nucleic acid makes an ideal target sequence because it is highly conserved among species. A single assay can therefore detect multiple mycoplasma targets. Other assays can further ta
rget ribosomal RNA (rRNA) or DNA (rDNA).
One drawback of NAT methods is the limited sample volume that can be input. The compendial growth method uses 10 mL of sample, but most amplification methods are limited to a starting volume of ?1 mL, which makes it difficult to compare their sensitivity with that of traditional methods. Also, these methods are often vulnerable to contamination of both nucleic acids and cellular debris that can cause false-positive results. Generally, strict workflow requirements and a high degree of training in molecular biology are required to run NAT methods efficiently.
Real-Time Detection: The presence or absence of mycoplasma can now be determined through RNA detection within four hours using the MilliPROBE kit from EMD Millipore. This assay starts with a representative sampling of up to 20 mL of a test article. Sample preparation is then coupled with Gen-Probe’s target capture and real-time TMA technology using the newly developed background reduction technology (BRT, Figure 3).
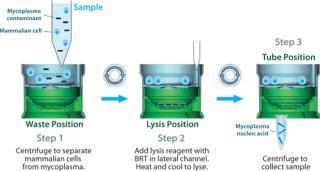
BRT protects the assay from false-positive results by ensuring that contaminating nucleic acids or mycoplasma cells that enter after the closed sample preparation procedure cannot be amplified or detected (Figure 4). The combined technologies of a closed, high-volume sample preparation and BRT protection provides for a robust assay with low false-positive rates. This allows it to be run without workflow controls typically used with other NAT tests (Figure 5).
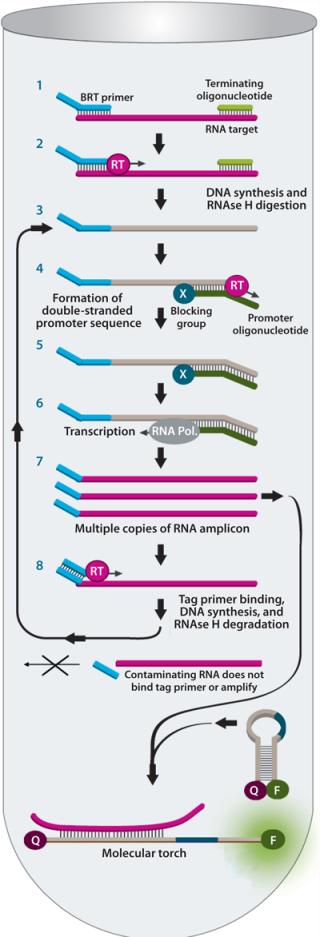

Mycoplasma testing is highly regulated, so new tests such as this nucleic acid amplification method first must be demonstrated to be comparable to the general growth-based method most commonly used in the United States. This standard requires users to demonstrate that their tests can detect <10 colony forming units per milliliter (cfu/mL) of mycoplasma without registering nonmycoplasma contaminants. The MilliPROBE system detects <1–10 cfu/mL for a panel of mycoplasmas and has been demonstrated not to detect a panel of other common bioreactor contaminants (see the “Performance Data” box). Improved Potency and Immunogenicity Assays
A second imperative outlined by PCAST was to shorten the time and increase reliability of reagent preparation for potency testing. The report suggested funding research to develop rapid methods for assessing the concentration of antigenic materials, which could circumvent the need for production of new antibodies and/or traditional immunological tests.
An important step in manufacture of many vaccines (including inactivated influenza vaccines) is testing product materials to determine their antigen concentration and determine whether this level of antigen can elicit an immune response. This potency testing can take on a number of different measures. Frequently, true potency measurement — testing the ability of a product to elicit a protective immune response in humans — cannot be carried out. Therefore, other related markers are used instead. In the case of influenza vaccines, the level of hemagglutin protein is often used as such a marker.
The hemagglutinin inhibition (HAI) method typically used for testing potency of inactivated or killed vaccines is straightforward and is not time consuming itself. However, the test reagents are sheep/goat/rabbit antibodies raised against a viral protein hemagglutinin, and it can take eight to 12 weeks to produce those antibodies (or longer if problems result). The process for doing so requires formal qualification, which takes another two to four weeks. According to the PCAST report, sheep antisera required in potency testing of inactivated vaccines for the 2009 H1N1 virus became available ten weeks after the reference virus was provide to the FDA by the Centers for Disease Control and Prevention (CDC). A further disadvantage of the HAI method is that it does not correlate directly with clinical outcomes: That is, the ability to hemagglutinate does not necessarily relate to protection in humans, which is recognized to vary within and among strains.
MILLIPROBE PERFORMANCE DATA FOR MYCOPLASMA ASSAY
The MilliPROBE system detects <1–10 cfu/mL for a panel of mycoplasmas and has been demonstrated not to detect a panel of other common bioreactor contaminants.
Result Type: Presence–absence result by RNA detection
Time to Result: ~4 hours
Sensitivity: ?10,000 mycoplasma rRNA verified with Acholeplasma laidlawii, Mycoplasma arginini, Mycoplasma gallisepticum, and Spiroplasma citri
Limit of Detection: <1–10 cfu/mL verified with A. laidlawii, M. arginini, Mycoplasma arthritidis, Mycoplasma fermentans, M. gallisepticum, Mycoplasma hominis, Mycoplasma hyorhinis, Mycoplasma orale, Mycoplasma pirum, Mycoplasma pneumoniae, Mycoplasma salivarium, Mycoplasma synoviae, and S. citri
Specificity: No cross-reactivity with 104 cfu of Acinetobacter baumannii, Bacillus cereus, Bacillus subtilis, Clostridium perfringens, Clostridium sporogenes, Corynebacterium jeikeium, Lactobacillus acidophilus, Lactobacillus plantarum, Micrococcus luteus, Propionibacterium acnes, Pseudomonas aeruginosa, Ralstonia pickettii, Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pneumoniae, and Chinese hamster ovary cell nucleic acids
Because neutralization measures the reduction in virus infectivity of antibodies and correlates with immunogenicity, it is considered to be an improvement over HAI methods. Although neutralization assays have potential for variability, strategies exist to mitigate the problem. Figure 5 demonstrates the accuracy that can be achieved by implementing modifications to the standard neutralization assay methodology that has existed for many years. In terms of these assays, the generally accepted wisdom says that they display inaccuracies of ±0.5 logs, which is equivalent to a percent inaccuracy from –50% to +200% relative to the known, true value of a sample undergoing assessment. Our relative potency assay design uses plate randomization and sample preparation techniques to reduce inaccuracies to between –20% and +16%, depending on the individual method. At that level of consistency, a validated neutralization assay enhances reliability and robustness of batch-release testing, resulting in higher levels of confi
dence. Accelerated Fill–Finish
A third area of focus identified by PCAST was the need to assemble a manufacturing network with sufficient capacity to rapidly fill vials, syringes, and sprayers required for delivery of vaccines for influenza and other viruses. This requires working with industry to adopt advanced manufacturing practices, modify existing facilities, or construct new facilities. The council encouraged the government, working closely with manufacturers, to undertake a comprehensive study within six months. This should quantitatively assess current fill–finish capacity so a plan could be developed to ensure adequate capacity for producing sufficient quantities of prefilled syringes, vials, and/or nasal sprayers to meet the national need.
All vaccines must be packaged for clinical use. The “fill–finish” step for inactivated influenza vaccines involves transferring bulk product into individual single-dose syringes and single or multidose vials used for inactivated influenza vaccines. Nasal sprayers are required for the live attenuated-virus vaccines. This can be a major rate-limiting step in the process of delivering needed products, especially under the compressed timelines associated with pandemics, when seasonal and pandemic vaccines are being produced simultaneously.
Options for driving greater capacity include construction of new facilities with more machines for sterile filling and finishing, but this is both cost prohibitive and time consuming. Many existing fill–finish facilities are relatively inflexible, with long change-out times and high operating and facility costs. Validation of cleaning and sterilization processes; of lengthy, labor intensive set-up; and validation and training of procedures to ensure process repeatability all contribute to creating bottlenecks when time to market is critical.
SINGLE-USE TECHNOLOGY FOR VACCINE MANUFACTURING BY GEORGE ADAMS, MANI KRISHNAN, AND PRIYABATA PATTNAIK
The single-use industry has witnessed high growth in protein/antibody applications. After trying to keep pace over the past decade, vendors are now turning their attention to new opportunities in vaccine applications. Worldwide vaccine sales in 2009 reached about US$10.4 billion, and annual sales are projected to double to $20.8 billion by 2015. Developing countries in Asia and Latin America manufacture as much as 86% of global vaccines by volume. So manufacturers are talking about made-to-configure rapid deployment of facilities, and engineering companies are responding with numerous concepts. Disposables will be very pertinent to deployment of such templates.
Therapeutic vaccines currently represent a negligible segment of the vaccine market (Figure 1). But a majority of the growth in vaccines will occur there. From $100 million in 2010, this is expected to reach $2 billion by 2015 — and almost $8 billion by 2025. Other areas for growth include influenza (as new strains develop) and pediatric vaccines for emerging markets. As populations continue to grow in India and China, for example, demand for the latter can only increase.
Vaccines are intended to induce as strong an immune response as possible, but immunogenicity is a problem for monoclonal antibodies (MAbs). The major emphasis in MAb manufacturing is on purity. MAbs are primarily therapeutics, whereas vaccines are traditionally prophylactic in nature — although as stated above, therapeutic applications for vaccines are growing. A key difference between the two is in the amount of dose given to each patient. MAbs can be half a gram per dose, whereas vaccines are measured in micrograms. So bioreactor volumes differ by an order of magnitude, with MAbs produced at 10,000-L and even 25,000-L scales whereas vaccines range 200–2,000 L. Disposables are perfect for that smaller scale.
After nearly three decades of industry experience, the MAb template is more or less well defined. We all know the nature and sequence of typical unit operations. But the broad range of vaccine product types makes it difficult to come up with the same kind of general manufacturing template. After talking to many customers, however, EMD Millipore is coming up with some general descriptions.
Case Study
Toxoid vaccines are commonly use to treat diphtheria, tetanus, and hepatitis. These toxins are produced by bacterial fermentation, with toxicity reduced or surpressed so that it will stimulate an immune response without harming patients. Media preparation and inocula propagation begin the fermentation process, which usually takes ~42 hours. For harvest and clarification, companies choose either centrifugation or microfiltration using open-channel hollow-fiber or cassette tangential-flow filtration (TFF), followed by a bioburden-reduction step using 0.45-µm or 0.22-µm membrane filters. Toxoidation follows, in which formalin is used to weaken or surpress toxicity. This unit operation can take as much as 40–42 days and requires intensive monitoring of sterility and pH testing. The resulting crude toxoid is concentrated 60–90× with ultrafiltration/diafiltration (UFDF) and then usually treated with ammonium sulfate for precipitation — a two-step purification process. Further concentration and formulation using UF/DF are performed before final filtration prior to filling in ampules, vials, or syringes.
Single-use mixers, filtration assemblies, and sterile connectors are available for media preparation, inocula propagation, and fermentation. Small assemblies are used for sampling and small-volume addition. Single-use bioreactors are not applicable because higher gas-exchange rates are required than they can typically provided.
Single-use mixers can be used to prepare sodium hydroxide as a clean-in-place (CIP) agent when a centrifuge needs cleaning. With hollow fibers or open-channel TFF microfiltration, disposable assemblies may help manage feed and retentate fluids. Single-use vendors offer a range of sterile filters for bioburden reduction. Formalin preparation for toxoidation also can be prepared with a single-use mixer, and disposable sampling products can be used for testing sterility and pH. Membrane cassettes and hollow-fiber technologies are both available for UF/DF. And single-use mixers are again relevant in preparation of ammonium sulfate for precipitation — as well as in formulation down the line.
Most toxoid products combine more than one vaccine into a dose. Single-use technology may be used to blend formulation materials together, and vendors are even developing disposable assemblies to be used for fill and finish (1,2).
References
1 Jenness E, Gupta V. Implementing a Single-Use Solution for Fill–Finish Manufacturing Operations. BioProcess Int. 9(5) 2011: S22–S26.
2 Riedman D, Martin J. A Case Study in Qualification of Single-Use Filling Manifolds for Particles and Endotoxins. BioProcess Int. 9(5) 2011: S28–S35.
George Adams (george.adams@merckgroup.com) is vaccine program manager and Asia/India/Japan market manager, and Mani Krishnan (mani.krishnan@merckgroup.com) is director of Mobius single-use processing systems at EMD Millipore, 80 Ashby Rd, Bedford, MA 01730; 1-781-533-6967; www.millipore.comPriyabrata Pattnai
k, PhD, is technical manager of the biomanufacturing sciences network at Merck Millipore in Singapore, 65-6403-5308;
This box is adapted from a presentation given by EMD Millipore at Interphex in New York, NY, March 2011 at a lunchtime session moderated by BPI’s editor in chief, S. Anne Montgomery.
Disposables: An alternative to building new facilities is adoption of single-use fill finish assemblies. We recently reported on the results of such an operation at a vaccine manufacturer (3). For this manufacturer, filling time was reduced from 24 to 10 hours; this increased the average number of vials filled per hour from 3,000 to 10,000 (Table 1). Overall, fill time was reduced from 36 to 12 hours per campaign. In the 85 million doses filled to date, there have been zero contaminations.Table 1:?Comparing traditional and Mobius single-use fill–finish process at a vaccine manufacturer

Adoption of single-use technologies can also provide advantages when manufacturing in far-flung locations. Facility and infrastructure requirements needed to support a disposables-based manufacturing process are considerably less than that needed for an all–stainless-steel facility. Additional advantages include lower capital requirements and higher flexibility, which enables more rapid batch change-overs.
With vaccine manufacturing operations increasingly being located in emerging markets, the ability to transfer technology and processes between and among locations is critical to maintaining product quality and speeding time to market. Use of replicable single-use systems accelerates start-up, reduces facility costs, and speeds technology transfer. This is accomplished by eliminating the need to develop and implement clean-in-place processes (by simplifying training) and eliminating the need to perform sensitive aseptic connections. To the Future
The global market for vaccines continues to be strong. New vaccine categories and the need for a more rapid response to public health emergencies present opportunities for growth. Vaccine manufacturers can position themselves for success through development and adoption of new technologies that will help deliver innovative products and accelerate time to market.
Author Details
Corresponding author George Adams is the vaccine program manager (george.adams@merckgroup.com); Dr. Helge Berg is biomanufacturing sciences manager; and Patrick McCarthy is a technology specialist in rapid microbiology at EMD Millipore; and Dr. Daniel Galbraith is chief scientific officer of BioOutsource Limited.
REFERENCES
1.) 2011.Human Vaccines: A Global Strategic Business Report, Global Industry Analysts, San Jose.
2.) President’s Council of Advisors on Science and Technology 2010. Report to the President on Reengineering the Influenza Vaccine Production Enterprise to Meet the Challenges of Pandemic Influenza, Executive Office of the President, Washington.
3.) Jeness, E, and V Gupta. 2011. Implementing a Single-Use Solution for Fill–Finish Manufacturing Operations. BioProcess Int. 9:S22-S26.