G -protein coupled receptors (GPCRs) represent a target superfamily linked to many disorders across all therapeutic areas. Although this target class has been historically treated by small molecules and peptides, antibodies can offer a number of advantages over such molecules by virtue of their specificity, dosing frequency, and restricted penetration. They also can provide other functional effects specifically mediated by the Fc region (ADCC and CDC) as well as different modalities such as those offered by bispecific and antibody drug conjugates.
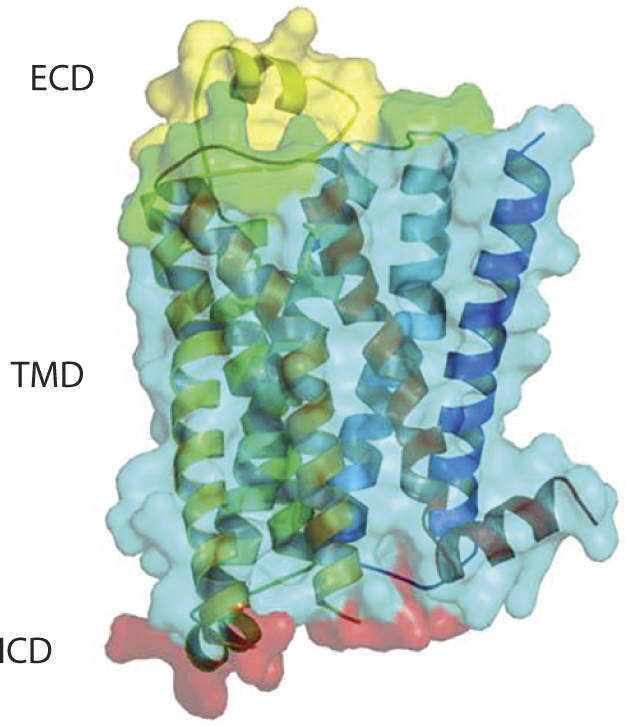
&bgr;1 AR Crystal structure depicting ECL2 in yellow
Similarly, a longer half-life in some instances (such as by FcRn-mediated recycling) can provide a therapeutic index that is more desirable than that of small molecules and peptides. In addition, antibodies can offer more diversity in pharmacological effects than provided by small molecules and peptides. This is because of the different nature of their extracellular interaction with a receptor compared with the interactions seen with small molecules and peptides, which often involve parts of the helices buried within the transmembrane regions..
Product Focus: Antibodies, GPCRs
Process Focus: Production, engineering, discovery
Who Should Read: Product development, analytical, clinical
Keywords: Stabilized receptors, phage display, expression systems, genetic engineering
Level: Advanced
However, the development of antibodies targeting GPCRs has lagged significantly behind antibodies for other drug target classes. The key challenge has been generation of sufficient stable GPCR antigens and immunogens that maintain the integrity of physiologically relevant epitopes. No robust or reproducible approach has been identified for either in vitro or in vivo methods. Binders are often obtained but not functional. Thus, sporadic success to date has been achieved only with receptors that have a higher than average stability and/or larger N terminal domains, such as subgroups of peptide receptors. The main issue to contend with is that GPCRs have a restricted extracellular surface where epitopes can be ligand sensitive (e.g., through binding that causes conformational changes in the receptor) and easily masked by long-chain detergents or lipids used for stabilization of purified receptors.
Here we summarize advances made in new approaches developed to address the challenges of antigen generation. We also review the antibody and biologics pipeline, with progress in the antibody space highlighted by some further interesting case studies. In Part 2, in BPI’s September issue, we review other biologics that have been evaluated or are in ongoing development for their use in targeting this important drug class of GPCRs
Enabling Technologies to Generate New Antigen Formats
We previously reviewed commonly used approaches to generate GPCR antigens (1). Although antibodies can be raised successfully and blocking antibodies have been described for a number of GPCRs (2), the success rate in delivering pharmacologically active antibodies is low. To provide correctly folded epitopes of extracellular domains or extracellular loops of GPCRs, presenting the receptor of choice embedded in membranes can
yield antibodies that recognize a biologically relevant conformation of the receptor (3).
That approach, however, generates a large pool of off-target antibodies and requires extensive screening. Another drawback is low expression levels — even in cells that over-express the target (e.g., by transfection technologies). However, a number of alternative emerging technologies are being implemented in monoclonal antibody (MAb) discovery for targeting multispanning membrane proteins. We describe a number of such examples below.
Novel Expression Host System: Tetragenetics has developed a system that could prove useful as an alternative for providing antigens in the form of whole cells or membrane vesicles for generating functional antibodies. The company applies Tetrahymena thermophilia to the over-expression of difficult-to-express membrane proteins (Tetraexpress technology). This technology can be used to formulate different formats of antigen preparations such as membranes, vesicles, and even soluble purified protein. The host system has been applied specifically to the cell-surface expression platform for ion channels (SionX technology). Tetrahymena is an interesting expression host for recombinant membrane proteins: It provides a large surface area and uses a significant proportion of its metabolism to produce membrane proteins. Therefore, the system can enable high-density expression of recombinant membrane proteins on the cell surface. Studies have shown that the target protein comprises up to 5% of the protein content in prepared membrane vesicles (4). This host system also is applicable for expression of GPCRs (personal communication).
Virus-like particles (VLPs) continue to show utility. For example, their incorporation into phage display selection strategies generated anti- CXCR2 MAbs that mediated efficient antibody-dependent cell cytoxicity and complement-dependent cell killing of CXCR2 over-expressing Chinese hamster ovary (CHO) cells using human serum with almost 100% cell death and IC50 values in the low nanomolar range (5). Effects of that study were specific for CXCR2- expressing cells. Also, a number of commercial efforts have been established that use liposomes (Integral Molecular), cell-free expression for the generation of proteoliposomes (Synthelis), and magnetic proteoliposomes (MSM Protein Technologies) in GPCR-MAb discovery.
Denatured GPCRs: The ideal antigen for therapeutic antibody drug discovery is correctly folded, purified protein. The purification of good quality membrane protein is still a very challenging field. For GPCRs, it is still very difficult to achieve milligram quantities of suitable antigen material. Researchers have described using denatured GPCRs as immunogens to raise highly specific antibodies (6). Although the generated antibodies appear to be specific and were able to show a positive response in cytofluorometric analysis, no functional data of these antibodies were presented. One disadvantage of this approach is that many GPCR epitopes in the native folded receptor will not be correctly preserved, thereby altering the diversity of the resulting antibodies.
Other research groups have used the advantage of the large soluble extracellular domains of some GPCRs to raise active antibodies. Those researchers implemented the extracellular domain of the glucagon receptor to generate MAbs that demonstrate in vivo activity (7). The human MAbs NPB112 effectively lowered glucose levels in diabetic animal models with mild and reversible hyperglucagonemia. Mice treated with NPB112 showed a significant improvement in the ability of insulin to suppress hepatic glucose production, so it might be a promising therapeutic modality for treatment of type 2 diabetes.
A different research group describe two antibodies (MAb1 and MAb23), where MAb1 inhibited the glucagon receptor by occluding a surface extending across the entire ligand cleft (8). The second antibody, MAb23, blocked glucagon binding and inhibited basal receptor activity indicating an inverse agonistic behavior.
Liposomes: Although purified GPCR protein can be unstable even when embedded in detergent micelles, reconstitution of the purified material into liposomes (which may include additional lipids and cholesterol) can provide purified receptor in a more stable format. Those additions result in an increased micelle size that can restrict the access of antibodies to GPCR epitopes. A few groups have published studies on functionally active antibodies raised from purified GPCRs. One publication describes generation of specific antibodies (by in vivo immunization using liposome-embedded adenosine A2A receptors) that recognize conformational epitopes (9). The Fab fragment of a functional antibody — which completely inhibited agonist binding — was used for cocrystallization. Structural analysis suggests that the receptor was locked into an inactive conformation by tight binding of the antibody to an intracellular domain of the receptor.
Studies focused on &bgr;
2
-adrenergic receptor (&bgr;2AR) have identified a camelid nanobody (Nb80) with G-protein-like behavior. To generate receptor-specific nanobodies, a llama was immunized with purified agonist-bound &bgr;2AR reconstituted at high density into phospholipid vesicles (10). Nb80 was then used as starting point to construct a library of mutants in which residues at the receptor-binding surface were randomized with conservative substitutions (11). The library was displayed on the surface of a yeast strain and subjected to a number of selection rounds using purified biotinylated &bgr;2AR. This process identified a nanobody (Nb6B9) that bound to the agonist conformation of the receptor (BI167107 ligand-occupied receptor) with affinities of 6.4 nM resulting in a 2.8 Å crystal structure complex of agonist–receptor–nanobody. However, this approach has been applied only to the creation of tools for cocrystallization. These antibodies are not suitable as therapeutics because they primarily target intracellular domains of the receptor.
Our group is now leveraging the Heptares StaR approach to generate antigens for GPCR antibody drug discovery, which has been exemplified by a proof-of-concept study based on the &bgr;1-adrenergic (&bgr;1AR) receptor (12). Although it has been postulated that a detergent-purified receptor is not a suitable agent for in vivo immunization because of detergent dissociation and subsequent receptor misfolding, we were able to obtain agonist MAbs against &bgr;1AR that recognized different epitopes on the receptor.
We designed &bgr;1AR StaR and used it as antigen (both DNA and protein) for in vivo immunization and subsequent hybridoma generation. Those MAbs enabled receptor signaling through G proteins in the absence of &bgr;-arrestin recruitment, indicating the potential for StaR proteins to elicit antibodies with highly selective pharmacology. The creation of a StaR allows purification in shorter chain detergents, thereby exposing larger receptor surface areas for antigen–antibody interaction. Increased receptor stability further improves receptor performance using in vivo immunization technologies. StaR proteins also are amenable to in vitro display platforms. A number of purified StaR antigens are being used in phage antibody library selection processes through a number of collaborations (unpublished data).
Monoclonal Antibodies Against GPCRs
By the end of 2012, 37 therapeutic antibodies had been approved and marketed around the world, generating sales of $64.5 billion in 2012 (13). Of the 10 best-selling drugs that year, six were MAbs, each with annual sales exceeding $5 billion. So far, however, only one GPCR-targeting antibody has been approved (Poteligeo mogamulizumab, an anti-CCR4 antibody approved in Japan), which reflects this central technical challenge of accessing reliable high-quality GPCR antigen.
We have published a review outlining case studies of antibodies currently in the clinic that target GPCRs — notably CCR5, CXCR4, CCR2, C5aR, and CCR4 (1). Based on information available through company websites, Thomson Pharma database, clinicaltrials.gov, and other sources, we summarized an updated overview of examples of MAbs currently in development (
Table 1), as well as identified further interesting case studies, as below.

Table 1: Examples of GPCR MAbs currently in development (NDRR = No development reported recently)
Anti-Bradykinin2 (DM-204) is a MAb targeting the bradykinin 2 receptor, which displays agonist behavior (14). Agonist antibodies are highly desired and considered to be very difficult to achieve. They offer several advantages over shorter-acting peptides or small molecules. DM-204 is a novel MAb to treat Type 2 diabetes (T2D) and some of its associated complications. In an independently conducted study, chronic administration of DM-204 prevented disease progression within an in vivo T2D model (www.diamedica.com/Product_Pipeline/dm-204). Treatment with DM-204 resulted in statistically significant lower HbA1c levels, fasting blood glucose levels, systolic blood-pressure levels, and total cholesterol levels — all of which are important factors in T2D disease management. Reduction in HbA1c is the critical endpoint measurement for regulatory approval relating to a T2D medication. In addition, about 70% of Type 2 diabetics are prescribed antihypertensive medications to control blood pressure and prevent heart disease and stroke. It follows that the antihypertensive and cholesterol-lowering ability of DM-204 may significantly reduce the need for such medication and protect from heart disease and stroke complications in T2D.
Anti-protease activated receptor (PAR2) belongs to a family of GPCRs that are activated upon proteolytic cleavage of the N terminus of the receptor, thereby activating the receptor signal cascade. So far, transgenic mouse studies have associated PAR2 activation with inflammation, pulmonary hypertension, and other pathophysiological conditions (15).
The role of PAR2 in inflammation continues to be investigated. PAR2 activation has been described as protective rather than proinflammatory such as in airway disorders (16). The current understanding is that some functions of PAR2 may indeed be protective in response to endogenous proteases, whereas others are detrimental and promote disease progression.
A small panel of PAR2 antibodies are commercially available, but only a few selective ones have shown in vivo effects. SAM11 is the most commonly used antibody (MAb IgG2a raised against N-terminal residues 37–50) and has recently demonstrated in vivo efficacy by attenuating collagen-induced arthritis in mice (17). In one study, researchers reported a panel of PAR2 antibodies that demonstrated good binding affinity to PAR2 with dissociation constants as low as 100 pM (18). Those fully human antibodies, identified through the HuCAL GOLD phage display library using a peptide-derived from the N-terminal region of human PAR2, bound to the N terminus of the receptor and thereby prevented proteolytic cleavage and activation of the receptor. The affinity-maturated IgG4 antibodies 2B and 2D were able to inhibit trypsin-induced intracellular calcium release, cytokine secretion, and relaxation of rat aortic rings (18). The antibodies also showed inhibition of the inflammatory swelling response in a mouse model of inflammation. Further antibodies have been described in patents by Amgen (US20130189281) and Regeneron (US20110059095), but to our knowledge none of those antibodies have entered clinical validation.
Anti-S1P3 (7H9): Sphingosine 1-phosphate (S1P) is a lipid-signalling molecule. It is generated by the phosphorylation of sphingosine by sphingosine kinase. Most of its effects are mediated by a family of five receptors (S1P1-5R). Effects mediated by S1P include proliferation, survival, and cytoskeletal rearrangement (19). Some studies have also indicated a protumorigenic role of S1P correlating with increased proliferation and cell survival (20).
Involvement in the processes of angiogenesis highlights the importance of this pathway in cancer progression (21). Neutralization of S1P has a potent tumor-suppressive effect (22). The effects in breast cancer cells are largely mediated by activation of S1P3 (23). S1P3 receptor also has been shown to mediate proinflammatory responses in a number of physiological conditions (24). Specific antagonism could provide effective protection against the progression of certain cancer types or types of inflammation. A recent publication characterizes a MAb, 7H9, against the S1P3 receptor (25). The antibody was raised by immunizing mice with S1P3 peptide. Immunocytochemistry confirmed specificity for S1P3 receptor, and 7H9 was able to inhibit S1P3-mediated arrestin translocation and receptor internalization. 7H9 showed clear inhibition of S1P-mediated calcium response in RH7777 transfected cells and partially inhibited the Gi coupled cAMP response of S1P. Furthermore, 7H9 ameliorates systemic inflammation in mice after LPS challenge. The MAb also inhibited development of breast tumor xenografts in mice with a fourfold increase in necrotic regions of the 7H9-treated tumors. Further developments will demonstrate whether such an antagonistic antibody against the S1P3 receptor will be beneficial and clinically relevant.
Anti-CGRP-R (AMG-334): Migraine is the most prevalent of neurological disorders and can affect patients throughout their lifetime. CGRP and its receptors are largely expressed in neurons and glia, both centrally and peripherally. Peripheral actions of CGRP appear to involve neurovascular inflammation, which is an important factor for migraine (26). CGRP effects in the brainstem also have a key role in the pathophysiology of migraine by resulting in neurogenic inflammation (27). Therefore, CGRP is involved in the pathophysiology of migraine both centrally and peripherally (28). Pain improvement can be achieved by antagonism in the periphery, the center, or both. Brain penetration may not be essential for analgesic properties of such antagonists.
Effects of one of the most potent endogenous vasodilators have been extensively described for CGRP. Based on its physiological role, however, some concerns have been raised regarding CGRP receptor (CGRP-R) inhibition (29). Concerns that CGRP-R antagonists cause vasoconstriction (thereby presenting cardiovascular safety risks) have so far not been confirmed. In vitro and in vivo studies have repeatedly shown that CGRP-R antagonists (small molecules and antibodies) do not have vasoconstrictor activity on coronary arteries (30). The first CGRP-R antagonist to be tested in humans (olcegepant) showed no effects on systemic hemodynamics (31). Therefore, cardiovascular safety concerns regarding CGRP-R antagonists have yet to be elucidated, but additional data are required.
Four distinct small-molecule CGRP-R antagonists have demonstrated proof of efficacy, but all were discontinued for a number of reasons (29). Currently, two small molecules are in clinical trials (NCT01445067 and NCT01613248). Because small-molecule CGRP-R antagonists have failed to reach commercialization, free CGRP and CGRP receptors have both been targeted using MAbs that can bind and neutralize biological activity (32).
Three MAbs against the CGRP peptide ligand are currently in clinical trials: LY2951742 (Eli Lilly), ALD403 (Alder Biopharmaceuticals), and LBR-101/PF0442g7429 (Labrys Biologicals). But only one MAb is targeting the CGRP receptor (AMG- 334). Amgen is developing AMG-334 for prevention of episodic migraine where AMG-334 (a human MAb) inhibits the receptor. Two phase 1b studies are testing the safety and pharmacokinetic (PK) profile of single- and multiple-ascending doses in healthy volunteers and in individuals with migraine. In summer 2013, a phase 1 study focusing on single-dose administration was terminated, but the parallel multiple-dose approach is currently ongoing (NCT01723514). This antibody has since progressed to phase 2 clinical trials to evaluate its efficacy and safety in migraine prevention (NCT01952574) and will be compared with placebo on the change from baseline in monthly migraine days. A second indication for AMG- 334 also has been submitted to a phase 1 study for assessment in women with hot flushes associated with menopause (NCT01890109).
Anti-CCR4 Mogamulizumab: We previously described the status of a defucosylated humanized IgG1 antagonist derived from the POTELLIGENT platform, a technology to produce antibodies with enhanced ADCC activity (developed exclusively by Kyowa Hakko Kirin) (1). In 2012, mogamulizumab was approved in Japan for treatment of relapsed or refractory adult T-cell leukemia-lymphoma (ATL). It is currently the first and only anti-GPCR antibody that has received marketing approval under the name Poteligeo as an injection therapy. Phase 2 development is currently ongoing for adult T-cell leukemia-lymphoma (ATL) and cutaneous T-cell lymphoma in the United States and for peripheral T-cell lymphoma in the United States and Europe. Further clinical trials have been initiated and are currently listed as in recruiting stage (clinical trials.gov). Recent published case studies describe some adverse effects during the treatment with mogamulizumab. Diffuse panbronchiolitis has been described during a therapy for relapsed adult T-cell leukemia and/or lymphoma (33). The intent is to raise awareness of this complication and the possible development of such autoimmune diseases that arise most likely as a consequence of targeting T-regulatory cells that express CCR4.
Another case study reported development of Stevens-Johnson Syndrome (SJS) during mogamulizumab treatment (34). Further clinical trials and the broader use of mogamulizumab will shed more light on the frequency of rare side effects in the future.
Ahead in Part Two
In BPI’s September issue, the second part of this article reviews other biologics — such as peptides and alternative scaffolds — that have been evaluated or are in ongoing development for their use in targeting this important drug class of GPCRs.
Author Details
Markus Koglin, PhD, is associate director, protein engineering; and corresponding author Catherine J. Hutchings is principal scientist, antibody alliance management and strategic partnering, at Heptares Therapeutics Ltd, BioPark, Broadwater Road, Welwyn Garden City, Herts AL7 3AX; 44-1707358698; fax 44-1707358640; cath.hutchings@heptares.com. The HEPTARES name, the logo, and STAR are trademarks of Heptares Therapeutics Ltd.