New methods and platforms for rapid development and production of effective subunit vaccines have become a 21st-century imperative. Not only is it important to rapidly express and produce a large number of antigens, but those antigens must be expressed and folded such that their effectiveness in preclinical studies is predictive of their potential effectiveness as vaccines. This task has created a bottleneck in vaccine development because recombinant protein expression is difficult and time-consuming, involving a large number of variables. Highly developed bacterial expression platforms are particularly suited to overcoming many obstacles because of rapid growth rates and a large number of expression tools that have been developed to meet these challenges (1).

Deciding which tools or combinations of tools will be effective in producing a particular antigen is often difficult. Primary amino-acid sequences reveal little information on how best to express a given antigen. What has been needed is a way to apply parallel processing to expression strain development so that multiple strategies could be accessed and tested simultaneously. The Pseudomonas fluorescens–based Pfēnex Expression Technology platform is a complete and flexible bacterial expression platform thanks to the development and application of a highly integrated, off-the-shelf toolbox of multiple expression plasmids and host-cell phenotypes (2).
Strain Engineering
By applying a parallel-processing, combinatorial-biology–based methodology to strain engineering, highly productive hosts can be constructed and subsequently identified by the application of high-throughput (HTP) analytical methods for expressed protein titer and activity. All the work — including strain construction, screening, and fermentation range-finding — is accomplished in about eight weeks. Using this approach, rapid engineering of strains for high-level vaccine antigen expression and production has become much more reliable and precise.
Figure 1 describes available expression strategies — including transcription (3) and translation signals as well as native P. fluorescens protein secretion signals (4) — and engineered hosts available through the platform that are used to produce more than a thousand empirically designed, completely unique expression strains. These tools can be seamlessly accessed to combine any expression scenario with any engineered host by 96-well electroporation. Expression plasmids combining the strategies listed in the figure and competent host strains are all “off-the-shelf” items. The plasmids themselves all use the same cloning strategy, which removes all operational barriers to effective target gene cloning. Once constructed, expression strains are grown in deep 96-well plates. The P. fluorescens system uses a defined medium without antibiotics for plasmid maintenance: Plasmids are maintained by auxotrophic complementation (5).
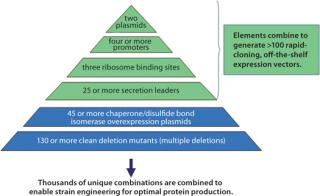
Figure 2 describes steps implemented in a laboratory to grow and assay expression strains for titer and (if possible) proper configuration of an expressed antigen or antigens. As an obligately aerobic organism, P. fluorescens is uniquely suited to growth by simple shaking in a temperature-controlled air chamber without oxygen supplementation. It lacks the physiological apparatus to switch to anaerobic growth, which produces organic acids (like Escherichia coli does) under microaerophilic conditions and limits a bacterial culture’s growth. A P. fluorescens organism continues growing as long as nutrients and oxygen are available. The result is accumulation of a significant amount of biomass: 30–50 A595 units. That enables production of a a significant amount of expressed protein with which to perform bioanalytical assays even at small scale.
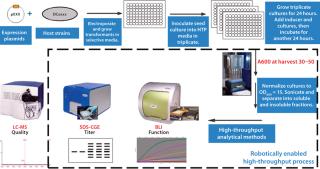
Typically we perform a two-tiered assay scheme. The first round of assays are HTP methods (at this scale we normally examine ~3,000 samples) designed to provide information on the titer of target protein expressed in the soluble cellular fraction (sodium dodecyl sulfate capillary gel electrophoresis, SDS-CGE), and possibly some information on its activity, if a protein–protein interaction assay can be implemented using biolayer interferometry. The boxed steps in Figure 2 are performed by a state-of-the-art sample preparation and analysis robot. With selected samples, the second tier of assays can include methods such as liquid chromatography with mass spectrometry (LC-MS) to further assess the quality of an expressed target protein. Because many proteins we express are fused to secretion-leader peptides, the LC-MS assay can provide information on the fidelity of leader cleavage by signal peptidase. All these steps — from receipt of a gene through completion of analyses and selection of strains to test in fermentation experiments — take about five weeks to complete.
Analysts at my company typically observe that 1–5% of constructed strains express the highest levels of target protein, and those titers are normally in a range of 100–2,000 mg/L in 96-well plates. At this point, strains are selected for fermentation “range-finding” experiments conducted in scaled-down fermentation equipment. First, at the 4-mL scale a design of experiments (DoE) routine is preformed on ≤10 selected strains. The DoE encompasses up to nine sets of experimental fermentation conditions to study variables such as temperature, pH, and induction conditions. In a further-down selection based on results at the 4-mL scale, two to four strains are studied in more conventional 1-L reactors to confirm the effectivenes
s of conditions identified at the 4-mL scale.
Those two rounds of fermentation studies together are accomplished in about one month, and they typically result in a three- to tenfold improvement over titers observed at the 96-well scale. Also, the volume of fermentation broth available from the 1-L reactors can be used to purify protein (grams in many cases) for preclinical studies. At this point a strain can be selected for cell banking and further downstream development. Thus, in little more than two months, 1,000 unique expression strains are constructed and evaluated, and a fermentation process is partially developed.
The object of these types of studies is to identify a production strain that expresses a sufficient titer of target protein to meet the demand for all clinical studies and eventual commercial supply. An important advantage of this rapid and precise development cycle is that in one round of development all supply demands can be met from a single strain with no need for rescreening or reengineering. Figure 3 shows the relationship between strain productivity and the number expression tools accessed through a strain-development and screening program. This relationship has been deduced from dozens of strain-screening studies performed by Pfenex scientists. The expectations of those studies were clear: Rapid identification of a highly productive strain that makes properly folded and “active” target protein. However, the curve in Figure 3 reveals an important corollary of this relationship between tools accessed and expression titer. More than one gene can be expressed while accessing fewer tools for each one so that the sum of tools used would be equal, and the outcome would be identification of less productive strains.
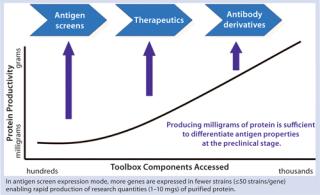
For certain desired outcomes, such results may be sufficient. For instance, if 25 potential antigens had been identified as vaccine candidates, they could each be expressed in 40 unique host strains with the reasonable expectation of identifying one or more that produce significant material (2–10 mg) to satisfy the needs of testing to differentiate among the candidates. To make purification and testing more efficient, a purification tag could be included in the coding sequence of expressed antigens. Once a smaller set of candidates was identified, a subsequent round of strain screening without a purification tag could identify the ultimate production strain or strains. Figure 4 provides an example of how such screening would be accomplished.

Our data show a snapshot of the expression of a single protein antigen from a set of 40 unique strains. This antigen contains one or more disulfide bonds, so it was expressed fused to a set of 10 different P. fluorescens-derived sec-type secretion leaders from the larger set described in Figure 1. Plasmids encoding those 10 were transformed into four host strains each, and 96-well expression experiments were performed as described in Figure 2. The SDS-CGE gel-like images in Figure 4 show that several strains express the antigen at reasonable levels. Depending on the position of the antigen in relation to host-cell proteins in the CGE traces, an expression level of at least 50 mg/L is required to clearly discern the expressed target. So in this experiment, expression by several strains would be more than sufficient to satisfy a need of 2–10 mg for preclinical, differentiating experiments. In this mode, the success rate for obtaining expression levels detectable by CGE is >80%.
Two Operating Modes
Recombinant protein expression has been a difficult and imprecise exercise due to the lack of a technology that allows parallel processing of expression strains, thus providing access to the large number of expression tools known to affect the amount and quality of expressed protein. The Pfenex platform overcomes that barrier. Operation of the technology in two distinct modes has been shown to provide strains suitable for clinical and commercial supply or to produce smaller amounts of material for preclinical testing to differentiate the candidates’ properties for progression of development. The significant advantage of this technology operating in either mode is the speed with which a production strain or strains can be developed. In the first case, this results in more rapid entry into clinical development. That significantly reduces the opportunity cost associated with laborious strain development. In the second case, it allows much more rapid and informed go–no-go decisions to be made on a larger set of candidate proteins.
Author Details
Charles H. Squires, PhD, is vice president for discovery and partnerships at Pfenex Inc., 5501 Oberlin Drive, San Diego, CA 92121, 858-352-4398, chs@pfenex.comwww.pfenex.com.
REFERENCES