Implementation of postlicensure process improvements in the biopharmaceutical industry can benefit patients and drug manufacturers alike. Here we demonstrate through a case study how a change to the cell culture medium and process can be taken from proof of concept through scale-up to demonstration of feasibility. We further illustrate the scope and complexity of implementing a change in commercial manufacturing to realize significant benefits such as increased production capacity over an existing legacy process.

Drug development is a complex process, and related business drivers typically shift through a drug’s life cycle. Before market licensure, the emphasis is on ensuring product safety and efficacy during clinical trials in addition to speed to market (Figure 1). The need to lock a process in as soon as possible limits opportunities for optimization. As a consequence, process optimization and capacity improvements are often postlaunch activities performed when the scope of market demand and the competitive business environment are both better understood. After a product is launched, related business drivers shift toward cost of goods (CoGs) and ensuring supply to patients while maintaining product quality. After launch and throughout a commercial production life cycle, process optimization is performed within the strict regulatory and quality frameworks of biopharmaceutical products, of course.
PRODUCT FOCUS: BIOTHERAPEUTICS
PROCESS FOCUS: PRODUCTION, MANUFACTURING
WHO SHOULD READ: PROCESS DEVELOPMENT, MANUFACTURING, QUALITY, AND ANALYTICAL
KEYWORDS: CELL CULTURE, MEDIA, PROCESS OPTIMIZATION, VALIDATION, REGULATORY AFFAIRS
LEVEL: INTERMEDIATE

Several approaches can help a company increase its plant output. A specific option is chosen to accommodate specific constraints. For example, if space in the facility allows, the size or number of production units (bioreactors) can be increased. Such changes to infrastructure and equipment often involve capital investment and entail regulatory submissions. Alternatively, the operating cell density (number of cells per unit volume) can be increased to increase a culture’s volumetric productivity (measured as product expressed per volume of culture per day).
Although we have successfully implemented those approaches in the past, other approaches could further increase capacity. For example (Figure 2), a cell culture environment can be improved by optimizing nutrient levels in the culture medium to increase cell specific productivity (product/cell/day). Medium components affect cell metabolism including protein synthesis, posttranslational modifications, and secretion. They can also affect the stability of expressed proteins after secretion into a culture medium.
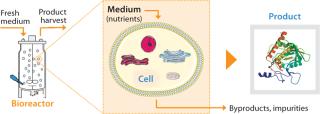
Many processes begin based on platform media types, but different production cell lines often have different nutritional requirements for optimal performance. Further, production cell lines may differ in their efficiency of nutrient uptake and use. Therefore, tailoring the medium composition to a specific cell source, to the mode of cultivation (e.g., batch, fed-batch, or perfusion), and to the specific process can significantly improve cell growth and productivity. Such an approach has the advantage of requiring no large capital investments or equipment changes.
Other approaches to capacity and cell-specific productivity increases (e.g., changes to a host cell line or expression construct) can require animal and/or clinical studies. But certain medium improvements would reduce regulatory submission requirements, which potentially reduces the time and costs associated with implementing such changes in commercial production. Such an approach has been successfully applied in the biotechnology industry before (1).
Media Enrichment As a Case StudyThe following describes an approach to increasing plant output with optimized levels of certain nutrients to increase the specific productivity of a culture. To facilitate commercial implementation, we focused on changes to the concentrations (but not the composition) of particular components of our cell culture medium. No new components were introduced. Our strategy was as follows:
-
Perform an extensive internal and external literature review.
-
Perform spent-media analyses to identify potentially limiting and/or depleted components.
-
Perform design of experiments (DoE) studies at bench scale using systems with much higher throughput than our bioreactors have. We developed and designed these to help us predict culture performance in a perfusion bioreactor (2) with titer being the primary output (Figure 3).
-
Verify a limited number of top leads using bench-scale perfusion bioreactors.
-
Select the top medium composition(s) and perform a full-length campaign run side by side with a control at scale, allowing for purification and product quality assessment.
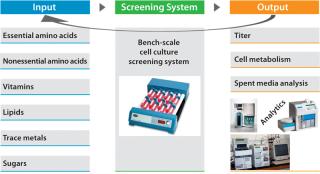
Our DoE work (Figure 3) used baby hamster kidney (BHK) production cells cultured in roller tubes (2). Results indicated that increasing the levels of a carbon source and a cofactor (enriching what was already part of the medium composition) would benefit productivity. We then followed with range-finding experiments on each ofthe two components, first in the higher-throughput roller-tube system and then in bench-scale bioreactors. The cofactor (component A) increased productivity by 17%, whereas the carbon source (component B) demonstrated a productivity gain of about 25% (Figure 4, LEFT). When we enriched both components together, product titer increased by ~35% (Figure 4, RIGHT).

Changes to media that increase cell-specific productivity can do so by affecting cellular metabolism. So it is important to verify that making those changes affects no impurity levels or product quality attributes (PQAs) such as glycosylation profiles. We addressed this by purifying our drug substance and subjecting it to the full cohort of product quality assessments. Further experiments demonstrated that both cell culture performance and PQAs remained comparable to those obtained when our standard medium was used (data not shown).
Proof-of-concept and feasibility studies showed that significant productivity gains could be achieved without impact to PQAs or to host-cell protein (HCP) or nucleic-acid impurity levels — and without a need for implementing major process or infrastructure changes. We also confirmed a lack of downstream effects complicating the purification process that can potentially result from increasing levels of both carbon source and cofactor (data not shown).
From Proof of Concept to ImplementationBefore a change can be implemented in commercial manufacturing, it must be validated, and performing a risk-assessment exercise first can be beneficial. The validation strategy should support evaluation of potential impacts on product quality and requirements for regulatory submission (as appropriate). The scope of this validation is determined by need and/or the scope of the associated regulatory submission.
A comparability protocol established for product and process characterization includes definitions of these elements: in-process testing, drug substance, drug product, extended characterization, stability, and acceptance criteria.
Experimental design considerations might include the scale at which validation is performed (in production or, if feasible, at pilot scale following justification of how well it represents production scale). The length of a campaign is another consideration. Such determinations often are made based on scientific knowledge of the change and experience with the production system. Decisions made should comply with the US Food and Drug Administration’s (FDA’s) and other guidances for industry (3, 4).
A regulatory submission may be required before a change can be implemented in commercial manufacturing. Whether it is necessary depends on the license range. It also depends on the nature of a given change, company experience, and so on. Such a regulatory submission package should include a scientific justification and be consistent with life-cycle management and continual improvement of process performance and product quality (5, 6). Other case-specific considerations include the submission type, country-specific regulations, the possibility of bundling with other changes, and business-specific considerations such as whether a change is worth pursuing from a CoG standpoint and strategic prioritizations within the company.
Implementation involves preparation and logistics activities that are typically driven by a change-control process covering areas such as personnel training, changes in equipment, and modifications to procedures. Affected documents can include process descriptions, technical registrations, and master data files. Warehousing, procurement, and quality control (raw material inspection and planning) are three in-house stakeholder groups heavily involved in preparations for implementation.
After implementation, follow-up should be continuous. Metrics are reported along the following three lines: postimplementation commitments (e.g., stability testing up to and/or beyond shelf life), routine process monitoring (to track potential long term effects of a given change), and pharmacovigilance (routine monitoring for adverse events).
Successful implementation of a process change depends on skillful project management from discovery through implementation, which in turn calls for a multifunctional effort relying on input and resources from many stakeholders. Specific levels of contribution (from different departments) change throughout the project phases (Figure 5) and can vary from one organization to another. Business “fit” is critical for reaping the benefits of any process improvement. For example, would the (improved commercial) process be expected to run for sufficient time to return significant benefits? And could the improved process serve other (e.g., next-generation) processes and products?

Postlaunch process improvements are a routine and important aspect of the biopharmaceutical manufacturing life cycle. Our small-scale experiments in varying media components showed good productivity improvement without affecting product quality. We determined that making a change would significantly increase our plant capacity and reduce operation costs. Such an approach could be applied generally toward other postlaunch improvements.
The media-improvement approach can complement other capacity enhancement strategies. Compared with many others, it may entail lower complexity and cost to implement in a current good manufacturing practice (CGMP) compliant production facility. It involves no genetic manipulations or changes to cell lines. And it requires no major changes to infrastructure or production processes. Implementing such promising results is a complex, multiyear program that involves numerous and diverse stakeholders across an organization along with strict oversight by regulatory bodies.
Author Details
Corresponding author Yuval Shimoni is principal engineer, and Venkatesh Srinivasan is director of manufacturing sciences at Bayer HealthCare LLC
, 800 Dwight Way, PO Box 1986, Berkeley, CA, 94710; 1-510-705-5775;