Risk of cell-culture contamination is a common concern whenever materials are added to or removed from a bioreactor. It is essential to maintain a sterile barrier and provide containment against intruding organisms during such operations. Many R&D and pilot-scale manufacturing tasks involve flexible, single-use processes with presterilized containment systems in nonclassified laboratory areas.
Here, we examine a process that requires substantial manipulation of a culture — first completely removing and later returning the culture to a bioreactor during media exchange — using individual presterilized, single-use bioprocessing components. Bags, sensors, and filters all needed to be assembled together and connected to the bioreactor beforehand (without laminar-flow hood) in a nonclassified laboratory. In addition, we tested a new method combining media exchange and clarification using a single filter sequentially in both operations.
PRODUCT FOCUS: M
PROCESS FOCUS: PRODUCTION
WHO SHOULD READ: PROCESS DEVELOPMENT, QA/QC, ANALYTICAL
KEYWORDS: BIOREACTOR, CULTURE MEDIA, SINGLE-USE
LEVEL: ADVANCED

CHO XM111-10 cells produce the recombinant enzyme secreted alkaline phosphatase (SEAP). Production of this protein is controlled by a promoter that is inhibited in the presence of tetracycline. Our production process with this cell line is divided in two phases:
-
growth phase performed with tetracycline-containing media; the objective is to allow cells to achieve desired cell density while inhibiting energy-consuming SEAP expression.
-
production phase, performed without tetracycline in the growth medium; the objective is to maximize production of SEAP.
To remove the tetracycline, a medium exchange must be performed between those two phases. The standard method in the Zurich University of Applied Sciences (Switzerland) laboratory is cell sedimentation in a cultivation system. After discarding old growth medium from the supernatant, settled cells are resuspended in fresh production medium. Substrate and oxygen limitations occurring during the relatively long sedimentation period (three to four hours) and loss of cells in the supernatant (up to 30%) are the disadvantages of this method.
We applied single-use systems from GE Healthcare to overcome those limitations and disadvantages. We connected a fully disposable crossflow filtration circuit to the disposable chamber of a single-use bioreactor system. The circuit consisted of a hollow-fiber cartridge; a recirculation loop with a recirculation bag; and additional bags for buffer, medium, and waste. Figure 1 shows a chart for medium exchange during our process and product separation with the same installation at the end of the production phase.
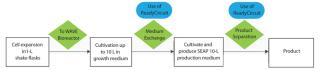
Table 1 shows systems used in the study. All consumables were delivered presterilized by gamma irradiation and were equipped with disposable aseptic connectors for sterile connection.
Table 1: Materials used in aseptic media exchange and protein harvest
Medium Preparation: We prepared the chemically defined CHO master culture and production media for the production run for SEAP using the cell line CHO XM 111-10 according to in-house and manufacturer’s (Cell Culture Technologies) protocols. For the maintenance culture in static culture systems (disposable T75-Flask), we used FMX-8 medium containing 2.5 mg L-1 tetracycline (for repression of SEAP production), 1.3 mg/L pyromycine, and 4 mg/L G-418 sulfate. For dynamic cultivation systems (shake flask and rocking bioreactor systems), we used the ChoMaster HP-1 growth medium (0.2% Pluronic F-68 and 2.5 mg/L tetracycline) and the ChoMaster HP-5 production medium (0.2% Pluronic F-68, no tetracycline addition). Pyromycine and G-418 sulfate were not added to HP-1 and HP-5 medium.
We prepared FMX-8 culture medium according to the above mentioned instructions (8-L kit) and supplemented it with tetracycline and sterile filtered in presterilized 1-L Schott flasks. After running a sterile test over 24 hours at room temperature (light protected), we stored the medium at 4 °C until use. Two 8-L kits of ChoMaster HP-1 medium were reconstituted one week before seed inoculum production in shake flasks and Cellbag cultivations. We sterile filtered 12 L of prepared HP-1 medium into a 20-L medium bag with a peristaltic pump. The medium bag and ReadyCircuit sterilizing-grade filter were connected with ReadyMate aseptic connectors. We sterile filtered the remaining 4 L into presterilized 1-L Schott flasks. A sterile test for both medium bag and Schott flasks took place over 24 hours in a dark, room-temperature environment. After visible inspection of the culture medium (no visible clouding), we stored the medium bag and Schott flasks at 4 °C until use.
As Figure 2 shows, we connected the medium bag to the Cellbag chamber. A feeding pump transferred 3 L of HP-1 medium into a Cellbag chamber one day before inoculation to equilibrate the optical pH sensors mounted on the Cellbag chamber’s floor.
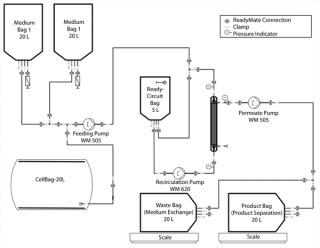
One day after inoculating the Cellbag chamber, we prepared the production medium (HP-5) according to working protocols. We prepared three 8-L kits and sterile filtered 20 L of the medium into a 20-L medium bag. We conducted a sterile test for 24 hours at room temperature (light protected), and stored the medium bag at 4 °C until use. The 20-L medium bag with production medium was connected to the Cellbag chamber with ReadyMate connectors (Figure 2).
Inoculum Preparation: At each handling step, we measured substrate and metabolite concentrations, viable-cell density, and viability because the media are chemically defined and require a sophisticated inoculum production strategy. Furthermore, the medium was tempered to 33–37 °C before use.
Starting with a static maintenance culture of CHO-XM 111-10 cells in FMX-8 medium, we pooled 10 T-flasks into a sterile bowl (each 15-mL cell suspension with about 1.2–1.5 × 106 cells/mL) and supplemented in total with 200 mL fresh HP-1 growth medium. After a sedimentation period of two hours at 37 °C and 7.5% CO2, the medium in the supernatant was removed, cells were resuspended and viable-cell density and viability were determined. We used that cell pool to inoculate two 500-mL disposable shake flasks with 0.8 × 106 cells/mL in 100 mL HP-1 growth medium (37 °C, 7.5% CO2, 120 rpm, 25-mm shaking diameter).
One day after inoculation of the shake flask, we determined viable-cell density and viability. We also inoculated two 1-L and one 500-mL disposable shake flasks (backup) with a cell density of 0.5 × 106 cells/mL in 200- and 100-mL HP-1 medium and incubated them as described above for two days. This inoculum was used three days before introduction to the Cellbag chamber to inoculate eight 1-L disposable shake flasks with 0.5 × 106 cells/mL in 150-mL culture volume (HP-1 medium) and incubated those at 37 °C, 7.5% CO2, 120 rpm, and 25-mm shaking diameter. After two days, we checked viable cell density, viability, and substrate as well as metabolite concentration in each 1-L shake flask, then diluted the cells with 70 mL additional HP-1 growth medium to ensure sufficient glucose concentration (>0.5 g/L) and exponential growth of CHO cells. Three days after inoculation of the 1-L shake flask, we measured viable cell density, viability, and substrate as well as metabolite concentration of each shake flask. We pooled two shake flasks at a time and added 150 mL fresh and tempered medium. The cells settled for two hours at 37 °C and 7.5% CO2. After removing the medium in the supernatant, we measured viable cell density and viability. We calculated the amount of cell suspension necessary to start the Cellbag cultivation at 0.8 × 106 cells/mL in 5-L culture volume.
Cultivation: We inoculated the Cellbag chamber at a viable cell density of 0.8 × 106 cells/mL in 5-L culture volume. Before this step, we equilibrated and calibrated the optical pH sensors with 3 L growth medium. The calculated amount of prepared cell suspension was sterile transferred into the Cellbag chamber under laminar flow, and the chamber was installed on the bioreactor platform. We adjusted the culture volume to 5 L with HP-1 growth medium out of the medium bag using the internal balance and feeding pump.
The aeration rate for the growth and production phases as well as the rocking angle was kept constant at 0.3 L/min (0.015-0.03 vvm) and 6°, respectively. We started the growth phase at a rocking rate of 14 rpm. We increased it to 21 rpm one day after inoculation, when cells were diluted with 5 L HP-1 growth medium to reach maximum culture volume. To prevent oxygen limitation, we increased (stepwise) the rocking rate (Figure 3).
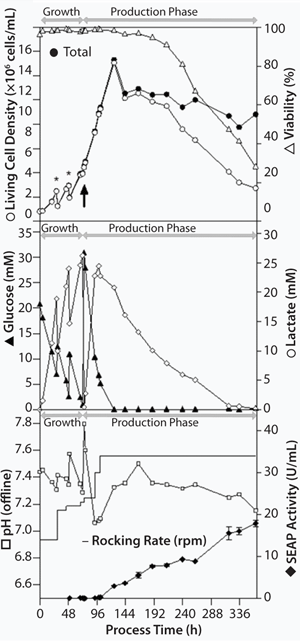
Figure 3:
Three days after inoculation, we exchanged the growth medium with production medium using the ReadyCircuit system to induce the production phase. For additional induction, we decreased the cultivation temperature from 37 °C to 31 °C one day after medium exchange and set the rocking rate to 34 rpm.
Growth and production phases were sampled once or twice daily, for which we withdrew 5 mL cell suspension from the Cellbag chamber. We used 2–3 mL of each sample to determine viable cell density, viability, average cell diameter, substrate, and metabolite concentration with the NucleoCounter NC-100, CedexHiRes, and BioProfile 100 plus devices. And we centrifuged two 1-mL samples for two minutes at 3,000g and stored the supernatant at –20 °C for SEAP analysis.
Medium Exchange and Product Separation: For medium exchange and product separation, we connected the Cellbag chamber (with the connected medium bags) to the ReadyCircuit flow path (Figure 2). The flow path consisted of a 5-L recirculation bag and a 0.37-m2 microfiltration hollow-fiber cartridge on the retentate side. On the permeate side, the flow path included connections to a waste bag for medium exchange and a product bag for product separation.
At the start of the process, medium was recirculated to remove air from the recirculation loop and calibrate the recirculation reservoir for a retentate volume of 2 L (for media exchange) and 1 L (for the clarification step). To exchange the medium, we filled the recirculation bag with cell suspension from the Cellbag chamber. We set the recirculation pump to process flow (3 Lpm). To start the filtration, we opened the permeate clamp and set the permeate pump to 180 mL/min to adjust a controlled permeate flow of ∼30 LMH. That decreases transmembrane pressure (TMP) and reduces the possibility of membrane pore blockage with debris.
While concentrating the cell suspension in the recirculation loop to 2 L, we turned on the feeding pump and set it to 180-mL/min until the Cellbag chamber was completely drained and the reservoir volume reached the calibrated volume of 2 L in the recirculation loop. We closed the clamp to the empty Cellbag chamber and opened clamps to medium bag 2 with the production medium (lacking tetracycline) to start the diafiltration after the concentration step. Diafiltration of three volumes (a total of 6 L) continued at the same 180-mL/min rate with a constant 2-L retentate volume to remove inhibitory metabolites and the product-expression inhibitor tetracycline. We monitored that diafiltration progress by weight of permeate (waste medium).
After the diafiltration step, the washed cells were pumped back into the Cellbag chamber, and the recirculation loop was purged four times with 1.5-L production medium to transfer residual cells into the chamber. We adjusted the volume in the bag to 10 L using the internal balance and the feeding pump.
At the end of the target production step, SEAP target protein separation was performed with the same recirculation loop and using the same filtration cartridge. The start permeate flux was reduced to 25 LMH to further reduce the risk of membrane blocking caused by the higher amounts of cell debris and proteins. For this step, a 10-fold concentration, followed by a diafiltration of four volumes (a total of 4 L of media) was performed continuously on the residual 1 L in the recirculation loop. During th
e filtration processes, we recorded pressure data and permeate volume and took samples for cell, metabolite and product analysis.
Cultivation: Starting from a viable cell density of 0.8 × 106 cells/mL in 5 L culture volume (96.3% viability), CHO cells grew up to a cell density of 2.48 × 106 cells/mL and a viability of 98.3% (growth rate of 0.039/hour, doubling time of 17.6 hours). We diulted them with 5 L HP-1 growth medium to a cell density of 1.25 × 106 cells/mL to reach maximum culture volume after 29 hours cultivation time. We diluted the cells a second time two days after inoculation to avoid substrate limitations (glucose) and metabolite inhibitions (lactate). Therefore, 4-L cell suspension (2.93 × 106 cells/mL, 98.7% viability) were aseptically removed out of the Cellbag chamber under laminar flow. We filled the chamber again to 10 L with remaining 2-L growth medium and additional 2-L production medium.
Three days after inoculation, we performed medium exchange from growth to production medium with a viable cell density of 3.9 × 106 cells/mL and 98.3% viability (average growth rate of 0.041/hour, doubling time of 16.9 hours).
Three days after medium exchange, the culture reached a maximum viable-cell density of 13.07 × 106 cells/mL with a 98.1% viability (Figure 3), and the stationary growth phase was achieved due to depletion of glucose concentration. In comparable cultivations with the sedimentation method for medium exchange, maximum cell densities of 5 × 106 cells/mL were typical (1). That means a 260% increase in cell density was achieved just by improving the medium-exchange method. Based on empirical knowledge, rocking rate was increased to 34 rpm and the temperature was lowered to 31 °C to boost SEAP productivity. Within 12 days of production, viable cell density declined with an average death rate of 0.005/hour. The system reached a viable cell density of 2.47 × 106 cells/mL and a viability of 28.0%.
Twenty-four hours after medium exchange, SEAP activity was detectable in the supernatant. It increased during production phase up to a maximum of 18 U/mL at the end of the process (Figure 3). During standard cultivations including the sedimentation method for medium exchange, a maximal SEAP activity of 10 U/mL is expected. That implies an enhancement of productivity of at least 80%.
Medium Exchange: During medium exchange, permeate flow could be kept nearly constant at 30 LMH over the complete process time (Figure 4). After concentrating the 10-L cell suspension down to 2 L in the ReadyCircuit bag (recirculation bag), we performed a diafiltration with ∼6 L fresh production medium to remove remaining tetracycline and growth-inhibitory metabolites such as lactate. The concentration step took 45 minutes, and whole process including diafiltration ended after 78 minutes. TMP was stable at 0.035 bar during the process.
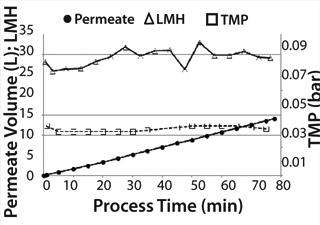
Cell density in the Cellbag chamber was 4 × 106 cells/mL with a viability of 98.3% before medium exchange. After concentrating the cell suspension, a cell density of 20.1 × 106 cells/mL was reached corresponding to a fivefold concentration.
Density of cells after they are returned to the Cellbag was 4.5 × 106 cells/mL. That slightly higher cell density could be caused by ongoing cell division during medium exchange and some measurement inaccuracy. Nevertheless, it could be postulated that this method of medium exchange provides a nearly 100% cell recovery. Due to the very short process time of <1.5 hours, the cells returned with a very high cell viability of 97.7% to the Cellbag chamber. The high cell recovery, high cell viability, and very good removal of lactate during medium exchange (Figure 5) resulted in high cell density amounts of about 13.3 × 106 cells/mL two days after medium exchange (Figure 3).
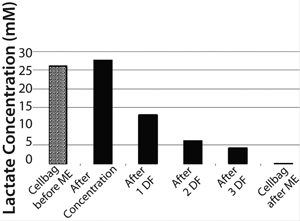
Figure 5:
Product separation at the end of the process (12 days after the inducing the enzyme expression) was performed with the same installation used for medium exchange. To increase the medium/buffer volume for diafiltration, we sterile filtered an additional 5 L phosphate-buffered saline, which was mixed with the residual product medium in the medium bag.
Before starting product separation, the hollow fiber cartridge (which was filled with production medium and unused for 12 days) was “reactivated” by filtering 1.5 L medium/buffer into the product bag at 25 LMH. We concentrated (10×) the 10-L cell suspension containing product in the recirculation bag, followed by a diafiltration (4 × 1L). The concentration step began with a controlled permeate flow of 25 LMH to prevent blocking the membrane caused by a higher concentration of cell debris due to lower cell viability of 28%. Permeate pressure decreased during concentration. To prevent a negative permeate pressure, we reduced (stepwise) the permeate pump power to a minimum flux of 20 LMH at the end of the concentration phase. After diafiltration, the permeate flow could be increased again by increasing the permeate pump power. TMP increased during the concentration step from 0.06 to 0.14 bar and was stable at this level during diafiltration (Figure 6). The concentration process took 65 minutes, and the whole process (concentration 10× and diafiltrate 4×) took 98 minutes.
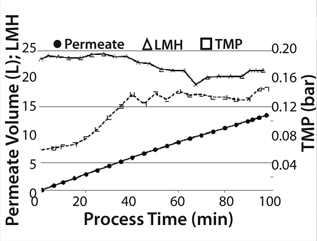
Figure 6:
During the filtration process, SEAP passed the filter membrane and reached the product bag. At the end of the concentration step, 79.2% of the total SEAP activity in the Cellbag chamber (170.3 kU = 100%) had been detected in the product bag. Diafiltration increased that recovery to 93.4%. We detected 96.1% of total SEAP activity from the Cellbag chamber in the retentate and permeate at the end of the process (Figure 7).
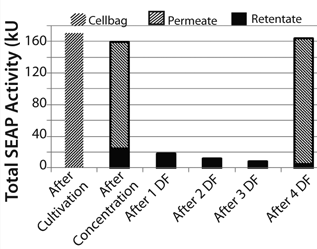
Figure 7:
Based on these results, we concluded that implementation of ReadyCircuit systems in our CHO process was very successful. High cell recovery and high viability was realized after medium exchange. Filtration performance during product separation was also very good, despite low initial cell viability. A 93.4% product recovery without optimization is an excellent result.
Not only could time for medium exchange be reduced from five hours to 1.5 hours with the ReadyCircuit system, but also the risk of contamination during production process could be minimized. Compared with the sedimentation method, the ReadyCircuit system provided a more homogeneous cell suspension during medium exchange. By doing so, substrate concentration gradients and limitations could be prevented.
Because of the very high cell recovery (nearly 100%) after medium exchange, a >2.5-fold higher cell density could be reached compared with the standard process (1). The higher cell density also resulted in an at least 80% higher productivity of SEAP. Reuse of the same filter cartridge for both process steps minimizes handling issues and improves overall process economy.
This ReadyCircuit application is a good choice for all two-phase processes requiring a medium exchange (e.g., transient transfection processes). Other applications could include cell concentration and preparation for cell banking.
Author Details
Sebastian Rothe is a research associate, Christoph Ries is a research associate, and Dieter Eibl, PhD is head of Cell Cultivation Techniques and Biochemical Engineering, all at Zürich University of Applied Sciences (ZHAW), School of Life Sciences and Facility Management, Institute of Biotechnology (IBT), Switzerland. Corresponding author Kieron Walsh is technical consultant for ReadyToProcess and filtration, and Wolfgang Hohenauer is sales development specialist, both at GE Healthcare Life Sciences, 14 Walkup Drive, Westborough MA 01581; 1-508-475-2045; fax 1-508-366-4751;