Implementing continuous process improvements is increasing in priority for the biopharmaceutical industry. Such implementation can be driven by product safety, purity, and stability enhancement opportunities as well as by cost-reduction pressures. Companies invest in projects to improve product quality assurance, safety, and yield as well as production efficiency (1). Such changes may come at any process stage, from early cell-growth methods through final-product packaging improvements. Examples include growth medium optimization, purification column operation optimization, and enhanced recovery during final filling (2). Continuous improvement and innovation projects often call for changes to licensed processes that require validation, but not necessarily animal or clinical studies.
PRODUCT FOCUS: RECOMBINANT PROTEINS
PROCESS FOCUS: PRODUCTION
WHO SHOULD READ: PROCESS DEVELOPMENT, ANALYTICAL, REGULATORY AFFAIRS, AND MANUFACTURING
KEYWORDS: PROCESS OPTIMIZATION, CELL CULTURE, SCALE-UP/SCALE-DOWN, MODEL SYSTEMS, PRODUCT QUALITY/COMPARABILITY
LEVEL: ADVANCED
Validation of cell culture process changes has been performed traditionally at scale, in a manufacturing setting, through the whole length of a campaign. However, it is infeasible for many companies to devote a full-scale production unit to a validation study because of potential impacts on production capacity output and/or supply risk, cost burden, manufacturing resource availability, and logistics complications (e.g., the need to segregate validation from commercial materials). Moreover, when a production unit is devoted to a validation study for a proposed process change, the resulting drug product manufactured often must not be released but rather kept on hold until stability and all predetermined acceptance criteria have been met and verified.

Putting product release at risk has multiple implications, including patient supply, operational, logistical, and financial consequences. All those can prohibit validation and lead to lost opportunities for potentially important production process improvements. Such problems affect many companies in the biopharmaceutical industry regardless of the scale of their production bioreactors (ranging from ~20 L to >20,000 L), modes of production (lasting from days, as in fed-batch mode, to months in the case of perfusion bioreactors), and the number of units available for manufacturing.
Given those constraints, establishing qualified scale-down platforms that need not comply with good manufacturing practices (GMPs) would facilitate continuous life-cycle process improvements (3). Validation of process changes thus could be performed without taking a production unit off line. That would allow for direct implementation of changes in commercial manufacturing without performing validation at scale. Such a scale-down system also would have utility in various characterization and process-development projects as well as in manufacturing-support investigations.
We focus here on the bioreactor unit operation and cell culture step because cell culture is rate-limiting for many biomanufacturing processes. A similar approach is needed for all unit operations, however, to allow product-quality verification — an essential aspect of qualification and process validation. In fact, ICH Q5B allows cell expansion under “pilot-plant or commercial-scale” for limit of in-vitro cell age determination for production (4). Moreover, scale-down purification systems that accurately reflect manufacturing process steps have been specifically endorsed by regulatory authorities. For example, virus-clearance studies in accordance with ICH-Q5A (5, 6) are performed using scale-down purification systems because it does not make sense to spike live virus into a unit located in a CGMP commercial manufacturing area. Such systems are now widely used (7). More recent guidance permits the use of commercially representative “laboratory or pilot-scale models” to estimate variability (6). We reasoned that the concept could be extended to include cell-culture/scale-down bioreactor systems in support of validation of process changes, so we provide here a “road map” to that end.
It may be impossible in many organizations to use commercial facilities for installation and operation of a scale-down platform to be qualified. In such cases, use of development-stage or pilot facilities should be permissible in alignment with internal quality assurance (QA) standards. For instance, Genentech has reported using good laboratory practice (GLP) compliant pilot-plant facilities equipped with 400-L bioreactors to aid validation programs for implementation at the 12,000-L scale (8). The company further demonstrated product comparability for its TNKase product. The Genentech authors ran their process at 400-L scale because of space constraints in the manufacturing facility. The material they generated was used to validate modifications made to the recovery process after phase 3 for implementation in a 12,000-L scale bioreactor process upon market launch.
Regulatory guidelines are typically written in general language (describing what needs to be done but not how to do it) because that allows for future applications of the most suitable, scientifically appropriate, advanced, and current methodologies. A recent FDA guidance for industry on process validation outlines a plan for equipment qualification and lists expectations for five items (6): studies/ tests, criteria, timing, responsibilities, and procedures for documenting and approving the qualifications. In line with those regulatory guidelines and expectations, we summarize below the principles and steps needed to develop and qualify a scale-down mammalian-cell bioreactor system to be used for validation studies. The concept is reduced to practice as a case study in part 2 of this article, which will appear in the June 2014 issue of BioProcess International.
The terms qualification and validation have specific definitions based on the context of the activities they encompass (as the “Definitions” box describes). One recent PDA technical report (11) provides a very comprehensive review, including the scopes of these activities that is consistent with current global regulatory guidance documents (12, 13). In accordance with GMP principles, process validation can be summarized as the collective activities “establishing scientific evidence that a process is capable of consistently delivering quality products” (6).
The goal of qualification (in the context of our discussion) is to confirm that components of a process — the scale-down bioreactor in this case — represent their production-scale counterparts and mimic their performance (within and/or beyond normal operation ranges, as discussed below). Using a qualified bioreactor and associated process equipment is critical for successful validation campaigns performed using those scale-down systems — and ultimately for successful implementation of process changes in manufacturing. The qualification plan detailed below applies the concepts of validation to a scale-down bioreactor that does not have to be located in a GMP production facility.
Our approach aligns with the US Food and Drug Administration’s (FDA’s) product life-cycle concept (6) and the expectations of “prospective” validation (based on a preplanned protocol). The study plan should specify criteria for concluding whether a process consistently makes acceptable quality product as well as provisions for addressing deviations from expected results and handling nonconforming data. Controlled conditions should be strictly adhered to (not just for cell culture or fermentation, but also all other functions including raw materials handling, assay methods, data integrity, and so on). The outcome of such a plan should be summarized in a qualification report to be approved and signed off by an organization’s quality unit.
Regulatory Input and Scope
We cannot overemphasize the need for early partnership with in-house quality and regulatory affairs departments. Once a draft (QA-approved) validation plan is in place, input regarding the feasibility of its adoption by regulatory agencies should be sought from the in-house regulatory affairs group. You need their agreement on your predetermined acceptance criteria. If necessary, you may seek scientific and regulatory input from key agencies as well. Qualification and approval of a scale-down system should be completed before its use in validation studies (intended to justify a process change to a licensed product).
Scope of the Qualification Plan
Regardless of the difference in volume between scale-down and production-scale systems, the scope of a qualification package comprises three aspects (6, 14): bioreactor design, performance, and quality. Design refers to specifications and geometry of a bioreactor and its associated parts, leading to equipment commissioning. Performance refers to response (time and extent) to input parameters and control, and to the capability of accurately targeting and maintaining desired outputs. Quality includes the system’s ability to make a product that meets predetermined quality attributes. It is essential to qualify the scale-down platform for each of those complementary aspects as explained below.
Bioreactor Design: Ideally, the scale-down bioreactor would be geometrically similar to its production-scale counterpart. This would entail a physical scale-down based on the production system’s specifications: e.g., the ratio of tank diameter to maximal liquid height (aspect), impeller and sparger types, and design and placement of probes and ancillary equipment within a vessel. However, a proportional duplication is not always possible because of equipment availability limitations. Thus, computational modeling is now often used to design scaled-down systems (15). The FDA also endorses computer-based and virtual simulations (6). Computational fluid dynamics (CFD) simulations for calculating energy dissipation rates (EDR) can be a useful scaling approach because EDR is a scalar quantity (unit: W/m3) that corresponds well to fluid hydrodynamic conditions (15).
Simulations help predict input values (e.g., for impeller power) that would yield similar physical forces to be experienced by cells when culture volumes are scaled down. Prediction capability is greatly facilitated when
- process and process sensitivity are well characterized
- process control strategies are well defined and consistent
- parameter interdependencies and linkage to responses are well defined and characterized
- bioreactor capabilities are well characterized.
Availability of such knowledge is usually an advantage of legacy products/processes for which relatively more process experience and documentation exist. Hydrodynamic and other simulations may be performed using model vessels (made of cheaper materials such as plastic or glass) before commissioning the (typically, stainless steel) real scale-down bioreactor. Simulations also may expand and be performed as part of performance qualification, which could lead to equipment redesigns for achieving representative performance.
Bioreactor Performance: Performance qualification (PQ) is an empirical process. The goal is to determine operational conditions of the scale-down system that will be needed to attain equivalent performance to conditions at production scale (16). This is challenging for scale-dependent parameters, as explained below. Volume-independent set points, by definition, should be identical for different scales. Examples of such parameters include temperature, pH, media composition, inoculation ratios, and feeding schedules (if applicable).
Scale-Dependent Parameters: Those parameters that depend on scale (volume) should be adjusted in proportion to vessel volume differences. Nevertheless, a linear scaling is not always accurate, especially when there are significant (or hard to avoid) design differences between scale-down and production-scale bioreactors. There may be additional issues.
Defining Key Terms
Process validation: Collection and evaluation of data — from the process design stage through commercial production — which establishes scientific evidence that a process is capable of consistently delivering quality product (6)
Process qualification: Proving and documenting that equipment or ancillary systems are properly installed, work correctly, and actually provide expected results; qualification is part of validation, but the qualification steps alone do not constitute process validation (7).
Process capability: Ability of a process to make a product that will fulfill its requirements; the concept of process capability also can be defined in statistical terms (10).
Analytical methods: Techniques of scientific analysis that support commercial batch release must follow CGMPs in 21 CFR parts 210 and 211 (6).
For example, oxygen control strategy should be identical between scales: sparger (and/or silicone-tubing aeration basket) design and location in the vessel, control set point, and gas mixtures used. However, because the efficiency of oxygen transfer and control is usually much better at reduced scales, operational settings (e.g., for agitation and sparge rates) will need to be adjusted to maintain equivalent oxygen transfer (kla), aeration, and dissolved oxygen (DO) control (Figure 1). Tip speed, Reynolds number, and power input per volume also can be used to determine equivalent agitation and sparging rates, as can oxygen uptake rate (OUR) and specific growth rate and titer of cultured cells. Other examples of scale-dependent parameters include total air flow, media delivery rates, and seeding procedures.
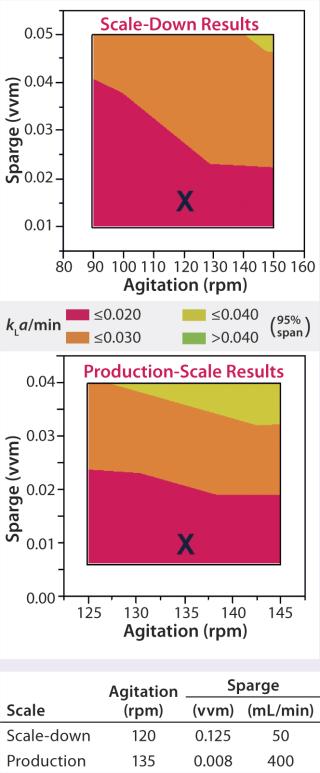
System Capability: For a bioreactor to be used in process validation, you must demonstrate that it is adequate for that purpose. Statistical tools (e.g., process capability index, CpK) can be very useful here. The scale-down model bioreactor thus should demonstrate less than or equal variability compared with the production bioreactor. Generally, a scale-down bioreactor that is better controlled and more sensitive to process changes than the large-scale system will help to ensure predictability of a changed process, particularly with regard to product quality on implementation of the defined change(s). Transcriptomics is an emerging tool that can also be used to assess scalability as demonstrated recently for mammalian cell perfusion systems (17).
Product Quality is the most important aspect of qualifying a scale-down bioreactor against the full-size production scale. As noted above, small differences in design (and performance, to a limited extent) may be unavoidable for reasons of technical and equipment availability. But product-quality equivalence between scales should not be compromised. To prevent surprises during implementation, the scale-down system should be no less sensitive to changes (that could affect process performance and/or product quality) than the full-scale system is. Quality assessment calls for an experimental plan consisting of the following steps, which all should be agreed upon with pertinent quality and regulatory stakeholders based on information available for the specific process:
- determining the output process and product attributes, assays, and acceptance criteria
- prioritizing process parameters (inputs) that will be tested
- defining operational ranges for each parameter to be assessed
- designing specific experiments for the different parameters tested.
Product quality attributes (PQAs) serve as the outputs of experiments to be performed (discussed below). The qualification program must use the same in-process and product-release specifications and tests as those performed in commercial manufacturing. These will include an array of analytical characterizations of the product bulk: product integrity, potency, glycosylation and other protein modifications known to alter product quality (e.g., strength, clearance, pharmacokinetics/pharmacodynamics, PK/PD), and stability. Because some tests require purified product in sufficient quantities, you should qualify a suitable purification scale-down system, as discussed below. PQA specifications are predetermined (usually in accordance with the desired function, mechanism of action, and known physiological effects of a given product).
Process Performance Attributes: Although they are not product attributes, cell metabolism and characteristics such as growth, viability, and titer — as well as residuals (host-cell proteins and nucleic acids, media impurities, and so on) — serve as important quality attributes. Specifications for process performance attributes could be set based on their known or potential impacts on PQAs.
Extended Characterization: Tests that are not a part of routine product release are usually optional in a qualification exercise. Focus may shift to specific attributes depending on the specific process changes being planned. For example, if a given change is known to potentially affect certain quality attributes, then it would be prudent to have those attributes tested as extended characterization outputs — (even) if they are not included in the in-process release tests. Extended characterization includes advanced technologies that may have not been available or ready to implement when a legacy process was initially locked in. Using such assays would increase confidence levels regarding the potential consequences of making a change.
Assays: Preferably, the methods and assays used to assess product quality will be identical for scale-down and commercial production processes. Following standard operating procedures (SOPs) whenever possible, inclusion of product release (quality control, QC) assay services is important to minimize differences. Likewise, sampling procedures, sample-collection devices, schedules, and processing (e.g., storage conditions) should be identical to both scales. Sampling time points, assays, and acceptance criteria should be determined in advance along with a plan for handling deviations.
Acceptance Criteria: You should determine your acceptance criteria before embarking on a qualification study. Ideally they would be agreed upon by all stakeholders in the company — notably the quality assurance (QA) group — and may be confirmed with pertinent international regulatory agencies (18). You should have a plan in place in case any criteria (including product specifications) are not met at any study phase. Determine your acceptance criteria in order of importance: first by scientific knowledge regarding impact to product quality, second by production specifications, and last by statistical distribution at set-point conditions.
Process Parameters Prioritization: Because it is impractical to validate every operational parameter for all possible conditions, a rational approach must be devised for prioritization of the most important parameters. Table 1 shows a common approach to defining parameters as critical, key, or neither, based on their effect on product when they are out of range/control (19, 20). Both critical and key process parameters (CPP and KPP, respectively) have a relatively narrow operation range. However, a parameter is deemed critical if it could affect the product and key if it affects only the process. Examples of parameters that are widely viewed as critical in cell culture and fermentation processes include temperature, pH, osmolality, nutrient concentrations, and DO levels. Parameters that are neither critical nor key do not affect the product or process and have broad ranges that are easy to control.
Table 1:

Assigning those labels relies on common professional knowledge in the industry, experience with a given process and product, and historical incidents. Risk-mitigation tools such as failure mode and effects analysis (FMEA) can be helpful in ranking parameters and usually require brainstorming sessions among subject matter experts (21). Incident and deviation reports (IRs and DRs, respectively) and historical documentation and company experience with a process will greatly assist in parameter classification. We recommend validating critical parameters by using experimental design tools as described below.
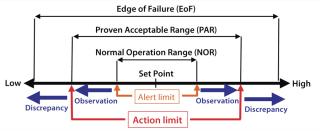
Operational Ranges: Operation ranges (Figure 2) for process parameters are classified as (19)
- normal operation range (NOR), that range within which a parameter typically fluctuates around its set point
- proven acceptable range (PAR), an operation range beyond or deviating from NOR that still yields acceptable product (depending on the specific parameter, exposure length and occurrence, this could result in an observation)
- edge of failure (EoF), an operation point beyond which the product can be affected. Such a discrepancy typically calls for an investigation (always for CPPs), resulting in an IR/DR that has to be closed out for the product to be released.
Experimental Design: The specifics and scope of your experimental design will be guided mostly by experience with the process within your company (22). The product-sponsor is expected to justify its approach. Thus, it is essential that stakeholders agree on the appropriate level of confidence and assess feasibility of performing the required work. We highly recommend use of statistical designs because they are science-based approaches that generate valuable data with expected confidence levels per number of experiments (23). Comprehensive experimental programs will be based on the following three elements: single factor and/or range-finding studies, DoE studies, and excursions.
Single-Factor Range Studies: Varying one factor at a time (OFAT) in range-finding studies allows identification of optimum levels for individual parameters. Optimum identification will help you determine input levels and ranges (center point, upper and lower ranges, and spacing between those settings) when moving on to the DoE stage. However, in-house data (if available) may allow you to move directly into DoE. Although an OFAT strategy can reveal effects of a specific parameter under set conditions, it cannot reveal the interactions among parameters and conditions. Another drawback to an OFAT approach is that it generally requires a large number of experimental conditions for high-confidence results (24).
Design of Experiments: DoE is a tool you are expected to use because its principles are in line with current regulatory expectations (6). DoE calls for planned testing and analysis, both of which rely on and generate process knowledge and scientific understanding, including interactions among multiple factors (24). We find it important that all parameters/inputs defined as critical be subjected to a DoE evaluation. Outputs should be ranked as well, with product-quality attributes ranking the highest.
A good design relies on both process experience and statistical tools. Experience is important to ensuring that the range of test variables is reasonable and that the increment between input levels is realistic. Use of statistical software is helpful for experimental design. Such software can be used for randomization and grouping (termed blocking) to avoid confounding effects of experimental cases, control setting, and replication. And of course, the software is used for data analysis after an experiment is complete or for augmenting the design as needed.
Excursions: Deviations of a certain magnitude from set input conditions that last a given time are called excursions. Reviewing incident history is a good approach to determining which parameters must be tested and the extent (magnitude and time) of your planned deviation for such testing. Excursion studies also can provide process capability information.
Downstream Process: Although we focus here on the approach to scaled-down bioreactor qualification, downstream process unit operations also must be scaled down accordingly and qualified to allow for the cohort of in-process and final-product quality testing. Qualification of proportionally scaled-down clarification, capture, and purification unit operations should be considered an integral part of every scaled-down manufacturing model.
Readiness
Facilities, Equipment, Raw Materials, and Personnel: All process components are expected to be controlled. A record-keeping system should be in place to ensure that SOPs are followed, including for facilities (e.g., maintenance, cleaning, environment, safety, and housekeeping materials); equipment (e.g., calibration and operation), raw materials control (e.g., receipt, sampling, storage, stability testing, and batch records), and personnel (e.g., job descriptions and training). Specifications, assays, test procedures, and approval processes also must be consistent with those in commercial manufacturing (6, 19).
Validation Approaches and Steps
Although a scale-down bioreactor can have multiple applications, our focus here is on appropriate practical steps needed to qualify one as a scaled-down validation platform for implementing process changes in production. The FDA has recently updated its 1987 guidance on process validation (6). As is characteristic of regulatory guidance documents and guidelines, it is written in a general language to allow the industry to apply tools that are current and scientifically the most appropriate to specific processes and products as time goes on. Early partnership with regulatory and quality functions within your organization — as well as with key pertinent international regulatory bodies — can enhance the probability of a successful validation campaign. ICH Q5E serves as a guideline for demonstrating product comparability after a process change is made (25). You should scientifically justify your validation approach and its specific components (e.g., parameters selection, ranges to be tested, and experimental design) and tailor them to your specific platform, process, and product.
Ahead in Part Two
Next month concludes our discussion with a case study describing scale-down of a perfusion bioreactor operating at a Bayer commercial manufacturing facility. Three multipart figures and a table will provide supporting data. Read Part 2 – Application – here
Author Details
Corresponding author Yuval Shimoni is principal engineer, and Venkatesh Srinivasan is director of manufacturing sciences at Bayer HealthCare LLC, 800 Dwight Way, PO Box 1986, Berkeley, CA, 94710; 1-510-705-5775; yuval.shimoni@bayer.com. Marc Jenne is at Bayer Technology Services in Leverkusen, Germany. Chetan Goudar is currently director of cell science and technology at Amgen Inc., 1 Amgen Center Drive, Thousand Oaks, CA 91320. Shimoni presented parts of this work at Cell Culture Engineering XIII in Scottsdale, AZ (22–27 April 2012).