Sponsors developing and manufacturing protein therapeutic products use a variety of analytical tests (e.g., cell-based potency and chromatographic assays) to assess quality attributes of their active ingredients and drug products. Those tests are used to assess product quality in a number of activities, including characterization, comparability, lot release, and confirmation product quality and stability.
Reference standards play a critical role in calibrating and confirming the suitability of such tests and in helping analysts to draw scientifically sound conclusions from data obtained. Different organizations create and use these standards in various ways, with approaches that are often unique to the type of standard material (e.g., in-house reference materials specific for certain product quality attribute testing or in-house primary standards).
In recent years, both experienced and relatively new manufacturers have shown increased interest in developing biopharmaceuticals. Consequently, there is significant value in capturing best practices for the manufacture, qualification, control, and maintenance of reference standards throughout a product’s life cycle. (Approaches other than those presented here can also be acceptable; so the content of this document is not binding regulatory guidance. Consult with your regulatory agency for specific reference standard strategies.)

To advance that goal, we summarize the findings of the California Separation Science Society (CASSS) Chemistry, Manufacturing, and Controls (CMC) Strategy Forum titled “Reference Standards for Therapeutic Proteins: Current Regulatory and Scientific Best Practices and Remaining Needs,” held in Gaithersburg, MD, on 15–16 July 2013 (1). Results of this forum have been collated with findings of previous CASSS reference standard meetings (2), two workshops held during the WCBP conferences 2012 and 2013 (3,4), and the conference “Reference Standards for Therapeutic Proteins: Their Relevance, Development, Qualification, and Replacement” (5). The latter was coorganized by the International Alliance for Biological Standardization (IABS), National Institute of Standards and Technology (NIST), National Institute of Allergy and Infectious Diseases (NIAID), and the US Food and Drug Administration (FDA) in September 2011. Together, these programs focused on selected reference standard topics, including
- initial qualification and life-cycle strategies from product development to postapproval maintenance
- potency assignment and potency stability monitoring
- assignment of content (mass and specific activity)
- critical operational aspects such as source material selection, configuration, and storage conditions
- regulatory expectations and experiences
- use of publicly available protein therapeutic standards and their role in biosimilars development.
Part 1 of this summary focuses on therapeutic protein reference standard life-cycle elements and practical implications of reference standards. Part 2 will focus on potency assignment for bioassay reference standards and the role of public reference standards in global harmonization of protein therapeutics.
Definitions Clarification
The “Definitions” box highlights some standards terms. For simplicity in this summary, both manufacturer’s in-house reference materials and international or national standards (as defined in ICH Q6B and ICH Q7) (6,7) all are referred to as reference standards. It should be noted that the term primary reference standard used here is distinct from a certified reference material (8), which can have the specific metrological meaning of a standard calibrated in Système International d’Unités (SI) units and traceable to the SI through a primary reference method. Thus the use of primary reference standard herein is distinct from a metrologist’s definition.
Reference Standard Life Cycle
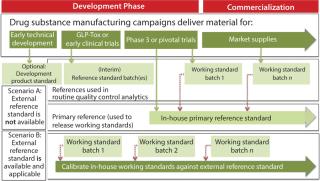
Markus Blümel of Novartis Pharma AG gave a detailed summary of best practices that have been identified in previous meetings and workshops on this topic, ending with the points that remained open from those discussions. The presentation highlighted definitions of various forms of reference standards (see sidebox). Figure 1 (from the presentation) summarizes the relationship of different types of reference standards evolving during development of a biopharmaceutical product. Depending on the applicability of an external reference standard (e.g., international standard or pharmacopoeial standard) either scenario A (in-house reference standard) or scenario B (external reference standard) would apply.
Sarah Kennett of FDA’s Center for Drug Evaluation and Research (CDER) presented on regulatory expectations for using reference standards during development and beyond. She provided details for using appropriate reference standards as critical to product development, licensure, and the continued life cycles of products. Kennett described some of the FDA Office of Biotechnology Products’ current thoughts and expectations regarding the development and use of in-house reference standards and reference standard protocols throughout a therapeutic protein product’s life cycle. She presented various case studies illustrating the challenges in maintaining the current standard and qualifying new standards.
Christoph Lindenthal of Roche Diagnostics GmbH presented on the transition from clinical to commercial reference standards. He described their course of development of requirements with respect to qualification, filing, and monitoring of a reference standard. He demonstrated that the approaches used during early development often differ significantly from what is expected for a reference standard used for a commercial product.
Definitions
In-House Primary Reference Standard: Per ICH Q7, reference standard primary is defined as “a substance that has been shown by an extensive set of analytical tests to be an authentic material that should be of high purity. This standard can be: (1) obtained from an officially recognized source, (2) prepared by independent synthesis, (3) obtained from existing production material of high purity, or (4) prepared by further purification of
existing production material.”
Specifically for biologics, rather than using an ultra-purified material as reference, ICH 6B states that “an appropriately characterized in-house primary reference material [should be] prepared from lot(s) representative of the production and clinical materials.”
In-House Secondary Reference Standard (also referred to as aWorking Standard): Appropriately characterized material prepared from representative clinical or commercial lot(s) prepared to support routine testing of product lots for quality control purposes, such as biological assays and physicochemical testing. It is always calibrated against a primary reference standard (either official or in-house).
In-House Interim Reference Standard: Appropriately characterized material prepared from representative clinical or used for quality control purposes during the development stage of a product. It is not compared to an official or primary reference standard, but it is established based on appropriate demonstration of its inherent characteristics.
Official Reference Standard: According to ICH Q7 definition, it is a primary reference standard obtained from an “officially-recognized source.” Typically it is established by a public agency (e.g. WHO), government (e.g. NIST, NIBSC), or compendia (e.g., USP, PhEur), and are officially recognized as standards by individual regulatory authorities.
Practical Implications of Reference Standards
John Ruesch of Pfizer Biotherapeutics presented a case study titled “Reference Standards: Overview and Strategy for Development to Commercialization.” He reviewed the critical role of reference standards in characterization, comparability, lot release, and confirmation of stability for therapeutic products. He showed how Pfizer BioTherapeutics Pharmaceutical Sciences has established a reference standard program that meets current regulatory guidance and expectations. Ruesch highlighted the current reference standard processes used during development and pointed out lessons learned along the way.
CMC Forum Series
The CMC Strategy Forum series provides a venue for biotechnology and biological product discussion. These meetings focus on relevant chemistry, manufacturing, and controls (CMC) issues throughout the lifecycle of such products and thereby foster collaborative technical and regulatory interaction. The Forum strives to share information with regulatory agencies to assist them in merging good scientific and regulatory practices. Outcomes of the Forum meetings are published in this peer-reviewed journal to help assure that biopharmaceutical products manufactured in a regulated environment will continue to be safe and efficacious. The CMC Strategy Forum is organized by CASSS, an International Separation Science Society (formerly the California Separation Science Society), and is supported by the US Food and Drug Administration (FDA).
Stacey Traviglia of Biogen Idec focused on improvements to commercial reference standards. She discussed improvements to the commercial reference standard program at her company. Traviglia illustrated their “lessons learned” using three case studies: batch-selection criteria for commercial programs, managing implementation of improvements to the qualification protocol, and primary reference standards.
Ashutosh Rao of CDER, FDA described FDA experiences with reference standard programs. He emphasized that general regulatory expectations are set forth in various regulatory guidance documents (e.g., ICH Q6B, Q2(R1), and Q7A) for reference standards of biologics. Those documents should be considered during development, licensure, and application of reference standards for therapeutic proteins. Rao presented case studies related to the appropriate versus deficient applications of reference standards, with the goal of sharing lessons learned during regulatory review of investigational and licensed therapeutic proteins.
Global Steering Committee for These Forums
Siddharth J. Advant (ImClone), John Dougherty (Eli Lilly and Company), Christopher Joneckis (CBER, FDA), Junichi Koga (Daiichi Sankyo Co., Ltd.), Steven Kozlowski (OBP, CDER, FDA), Rohin Mhatre (Biogen Idec Inc.), Anthony Mire-Sluis (Amgen, Inc.), Wassim Nashabeh (Genentech, a Member of the Roche Group), Ilona Reischl (BASG/AGES, Austria), Anthony Ridgway(Health Canada), Nadine Ritter (Global Biotech Experts, LLC); Mark Schenerman (MedImmune), Thomas Schreitmueller (F. Hoffmann-La Roche, Ltd.), Karin Sewerin (BioTech Development AB)
Panel Discussion
Concluding the life-cycle session was a panel discussion hosted by Markus Blümel, Manon Dubé (Health Canada), Sarah Kennett, and Christoph Lindenthal. (Discussions held in these sessions specific to potency standards will be included in Part 2). The “Practical Implications” session panel discussion was hosted by Manon Dubé of Health Canada, Ashutosh Rao, John Ruesch, Stacey Traviglia, Ramji Krishnan of Bristol- Myers Squibb, and Mikhail Ovanesov of FDA’s Center for Biologics Evaluation and Research (CBER). In each panel, comments and questions were fielded from panel members and attendees, guided under specific headings to facilitate the interactive discussions.
Establishing and Using Reference Standards: Speakers and attendees agreed that reference standards are essential in quality control (QC) testing of protein therapeutic products (e.g., to ensure the accuracy of results or to monitor assay performance). Quality attributes such as potency, purity, and identity of active ingredients or drug products are usually assessed during lot release or stability programs or through additional characterization testing. Reference standards are key elements in a control strategy. They ensure continuity of product quality, stability, and comparability throughout product development and commercial manufacturing.
Kennett of the FDA stated that regulatory authorities require that reference standards be suitable for their intended purposes, well characterized, qualified, and stable. She strongly recommended that appropriate protocols for manufacture and qualification of reference standards be in place. However, she noted that limited information regarding those expectations has been provided in published guidance documents.
Common Life-Cycle Elements: Kennett maintained that carefully considering and implementing many aspects of a reference standard program early in development will aid in the successful transition to licensure and support entry into different phases of clinical development. And Rao of the FDA emphasized a life-cycle approach for use and management of reference standards, reminding the audience that managing a reference standard program is a current good manufacturing practice (CGMP) expectation for licensed products. Blümel of Novartis summarized current best practices for a life-cycle concept in the preparation, qualification, control, and maintenance of a manufacturer’s in-house reference standard, as elaborated in several workshops over the past years.
Yet different organizations create and use reference standards in different and unique ways during development, as noted by Rausch of Pfizer. He demonstrated how his company’s approach is aligned with industry best practices, thereby illustrating the guiding principles critical in developing current reference standard approaches:
- Minimize the number of reference standards during development. Maintenance, support, and management of reference standards over a large portfolio of products takes a large number of people and add to the overall cost of development.
- Provide assurance that cell line and process development groups are aligned and able to support the minimal reference standard approach.
- Maximize implementation of heightened analytical characterization data gained on reference stand
ards for use in filings. - Remember that reference standards are the bridge back to clinical data. Always use them during comparability exercises during development.
In an overview of the life-cycle strategies at Roche Diagnostics, Lindenthal said that the first reference standard is typically filed and assessed for suitability before the start of clinical production. It comes from either a representative batch — such as good laboratory practice (GLP) toxicity or engineering batch — or from the first GMP batch. At this stage of development, manufacturers may have little data for shelf life claims and extensions and comparatively little knowledge of the product.
Manufacturing changes during development can trigger discussions on whether a new reference standard must be qualified and what potency value must be assigned (different values could be assigned when justified). For commercialization, a reference standard must be fully qualified and represent the commercial process. It also must provide a link between early and later development. For licensure, within the marketing application there must be a well-defined qualification program that includes a characterization strategy. The program also should include a strategy to demonstrate postapproval stability and/or trending to support continued suitability of the reference standard and shelf-life extensions. In addition, the postapproval reference standard qualification program must be defined.
A Two-Tiered Strategy for Reference Standards: Kennett emphasized that the goal should be to have a two-tiered system — a primary and a secondary (working) reference standard — at time of licensure. ICH Q6B recommends this two-tiered approach in reference standard programs for biopharmaceutical products: “In-house working reference material(s) used in the testing of production lots should be calibrated against primary reference material,” where the primary is either the well-characterized in-house reference standard or where available and appropriate, an international or national standard. Figure 1 illustrates how a two-tiered concept typically evolves during product development. As Traviglia of Biogen Idec showed in the cases studies she presented, implementation of a two-tiered strategy using primary and working reference standards — with careful consideration of batch selection and use of an appropriate qualification protocol — can all work to minimize drift over the lifetime of a commercial program when moving from one reference standard to another.
What is a current strategy to assess suitability of a reference standard for its purpose? It depends highly on its use(s) — e.g., method system suitability, calculation of a quantitative (or reportable) result — as well as the time in development when it is made. Quantitative use of a reference standard requires rigorous assessment of the “true” value of a material, or (as for potency) the most accurate value should be established and statistically justified. When the standard is used solely for comparison (e.g., “conforms to standard”), representative variants or process impurities should be present, depending on how the comparison is evaluated. If a standard is used solely for method system suitability (i.e., not used to generate a reportable test result), then rigorous quantitative assessment may not be necessary.
How do you address formulation and concentration differences among standards and test samples? As seen in Figure 1, a drug substance is typically suitable to serve as both drug substance and drug product reference standard. However, a drug product might be suitable, depending on formulation and other factors. If differences do exist between drug substance and drug product, manufacturers must ensure that analytical methods aren’t influenced by those differences (e.g., dilution for aggregates, interference by excipients). Attempts should be made to ensure that sample preparation is similar for the reference standard and for a sample to be analyzed. Alternatively, a separate control sample with the same formulation as the test sample could be used. Method qualification should determine whether dilution with buffers has influenced the accuracy of results.
What is the current strategy for selecting representative material used as the first reference standard or as primary reference standard? Early (interim) reference standards should reflect the clinical manufacturing process; primary (final) reference standards should represent the commercial process and have attributes linked to the clinically qualified material. Selecting material near the center of attribute limits might be preferable because it helps prevent drift to the extremes, but it may be not essential, depending on the intended use of the reference standard.
The primary reference standard is usually created at the latest in phase 3 or late-stage development. However, commercial material representative of the clinical product also may serve as the primary reference standard. In either case, sufficient material should be available to have enough primary standard to use for an extended period of time.
What is the acceptability of pooling lots for reference standards? Pooling multiple lots for reference standard manufacture may be possible. Pooling might be needed to ensure that lot size is sufficiently large or when the reference standard must represent properties that vary from lot to lot — when it represents an expected profile or ensures that an attribute is at an acceptable level (e.g., when used for system suitability). But all lots used for pooling should meet specifications, and production, testing, and stability information about those lots should be provided in the qualification.
How do you create a reference standard with limited lots during development? One audience member asked, “If a sponsor has completed pivotal trials using limited number of lots, can any of those lots be used as the primary standard without extended characterization, because that lot is representative of the clinical trial material?” In a typical product development plan, it is unlikely that early material would be selected for the late-phase primary or working standard. But even if it were justified, there is still the need to thoroughly assess that material to establish what are the clinically-qualified extended characteristics. The sponsor must also demonstrate that the material is suitable for all of its uses (e.g., QC assays, comparability). Moreover, extended characterization generates data to allow you to monitor stability and determine at a later time point whether a new primary standard is needed.
During development, how do you track attributes over time and across subsequent standards? Control data and day-to-day data from QC testing are often used to assess stability of a working standard. Using control samples, comparing historical data (e.g., chromatograms), and monitoring trends — particularly when associated with each switch in standards — can maintain a link between standards, even if the actual material has been used up. Also, if a working standard or control is being used regularly for QC testing, and the analytical data obtained are monitored, then a systematic and predefined investigation for any trend could replace a formal stability program. You should review each change in reference standard made during development and assess how attributes might have changed over time. Documentation associated with tracking and trending of changes in reference standards should be part of investigational new drug (IND) update filings, especially before phase 3.
Characterization and Qualification Elements during Development: Kennett stated that the qualification of reference standards must demonstrate suitability for intended use, which frequently goes beyond serving as a comparator lot for release and stability testing (e.g., system sui
tability). The level of characterization for qualification must be justified depending on how and with which tests the standard will be used. The rigor of qualifying interim reference standards might be limited at early development stages because neither the process nor the QC methods will be fully validated. However, even at phase 1–2, methods used for qualification of reference standards must be suitable for intended use (9). For early development, most participants indicated that they do not use a two-tiered system (primary and working standard) and some do not use formal protocols for replacing interim reference standards.
What is the level of characterization for a primary reference standard? Is there a trend toward the use of “state-of-the art” assays? Analytical characterization is usually expected to be extensive. Typically, the primary reference standards are the most “characterized” materials presented in marketing applications, often used in the 3.2.S.3.1. Elucidation of Structure section (10). It is expected that characterization of reference standards will use both established and state-of-the-art analytical methods (e.g., to assess higher-order structures). If appropriate techniques are not all available in-house, contract testing organizations (CTOs) may be used. However, as was noted in discussions, because these data are critical, the quality practices used by CTOs in performing tests should be suitable to assure their accuracy and reliability.
Do reference standard qualification protocols need acceptance criteria, and if so, what types? It depends on the standard’s intended purpose, the nature of each method used in the protocol, and the stage of development. Some sponsors may use “report result” in a reference standard protocol for certain methods, or in early development. However, regulators may request acceptance criteria, particularly late in development. That is when clinical data defining the quality, safety, and efficacy of the product will be available, and the tests required to characterize it have either been validated (e.g., lot-release and stability assays) or qualified (e.g., assays used only in characterization or comparability studies). Some methods can have rather broad acceptance criteria based on inherent assay performance or critically of the attribute. If using a release test, you might be able to use specification test limits in early development. But later you should consider tightening the limits to ensure that new lots provide a more centered reference standard.
Should the qualification criteria for a secondary/working standard be identical to those for the primary standard? The qualification criteria depend on the use(s) of the working standard. For some intended use(s) (e.g., qualitative comparative identity) a working standard could be less characterized and/or have wider acceptance criteria than the primary reference standard. For the relevant quality attributes, the same degree of rigor should be applied as for the primary reference standard.
Another view was that a working standard is usually prepared from a commercial lot, which was obtained from a validated manufacturing process and tested with validated test methods. Therefore highly rigorous qualification may not have to be repeated. Whatever approach is used for qualifying working standards, rationale for the proposed limits should be provided and justified.
What type of assay reporting should be done for assessing glycosylation profiles? “Conforms to reference” is deemed most appropriate for test samples. Simply using “report results” provides no assurance of control or consistency of heterogeneous species. But this raises challenges in determining how to define the expected pattern of glycoforms for reference standards against which conformance is assessed. It is necessary to define what is to be reported as a conforming profile, such as the pattern of species, the number of total species, and (in some cases) the relative abundance of each. The justification should include the quantitative and qualitative capabilities of the method(s) of measuring glycoforms, demonstrated process capability, stage of product development, and potential risk to product quality.
How do I switch to a two-tiered system? The first primary reference standard should be compared with clinical material using methods of suitable reproducibility and predefined acceptance criteria. Lots used for working standards should be representative of product and compared with the primary reference standard. Comparing the performance of working standard with that of primary standard might also justify situations in which it is fit for purposes that differ from the role of the primary reference standard.
How often should I change primary standards? Replacements of the primary standard should be kept to the absolute minimum. It requires extensive testing effort and copious amounts of data to qualify a replacement primary standard. So it is strongly recommended to use a working standard for routine tests to limit usage of the primary standard and therewith extend its period of availability as long as feasible.
What information regarding reference standards should be submitted in an IND or BLA? An IND should include a brief description of the source, manufacture, and characterization of a reference standard. It should also describe the analytical methods used to characterize the reference standard and include justification for the tests used. The marketing application should include the proposed use(s) of the standard with a thorough predefined protocol that includes justification of acceptance criteria. Using the ICH CTD document, 3.2. S.5 “Reference Standards or Materials” should contain the majority of information on reference standards (10). Many participants also noted that they place characterization data obtained from the reference standard in 3.2. S.3.1. “Elucidation of Structure.”
Test results should be clearly presented (e.g., representative and high-quality chromatograms). Distinct reference standards (e.g., for specific product or process-related impurities) may be required beyond the primary reference standard and should be described. If an international or pharmacopoeial standard is available, then data should be provided on comparison/calibration. Stability data should be provided in a marketing application, including the proposed stability monitoring concept. Attendees indicated that a standard operating procedure (SOP) is often developed within the quality management system for developing and replacing commercial reference standards. In S.5., distinct reference standards (e.g., for specific product or process-related impurities such as for HCP assays) may be required and should be described.
In the United States, including qualification protocols for both primary and working reference standards in a license application should provide regulatory relief when subsequent reference standards need to be qualified. That way, if a protocol is approved, then each new reference standard qualified according to that approved protocol can be described in an annual report. The same applies in Canada, with exceptions outlined in Health Canada’s guidance document (11).
What is the current strategy for replacing a primary reference standard after approval for commercialization? A new reference standard is not required simply because a change is made to a manufacturing process. If a process change is made that significantly alters a protein’s attributes, that then influences the use of a reference standard (e.g., impurity profile for mass determination, system suitability, or potency value), or the link to original clinical studies is lost (e.g., a new clinical study is undertaken), then a replacement primary standard should be manufactured. If a reference standard is starting t
o degrade enough to warrant replacement or inventory is getting too low, then a replacement should be considered.
Over time, tests that are part of an approved qualification protocol might change. Can a method change be addressed in the qualification protocol within a marketing application, as a prior approval supplement (PAS)? A qualification protocol in a marketing application could contain a forward-looking statement that methods may change, with a commitment to conducting appropriate method bridging studies to demonstrate that the new methods are equally good or better than the prior methods for the same intended uses. When filing a new assay (e.g., change of potency assay) as a PAS, including that assay in a revised reference standard protocol at the same time may be a simple way to update the approved protocol. Alternatively, a revised protocol could be submitted as a separate PAS well in advance of the qualification of a new standard.
Should all qualification tests for a reference standard be included in its stability monitoring program? The stability of all reference standards should be monitored in a predefined program with appropriate acceptance criteria. The set-up of the monitoring program can be based on information obtained during development (real-time and accelerated data, formulation information) and prior knowledge. A trending program should then be in place for monitoring stability.
At the IND stage, stability data will be limited, but material should be stored in a manner that prevents degradation (e.g., frozen). Comparing the primary reference standard to itself as part of a stability study is not of much value for quantitative assessments, so more qualitative data also should be included (e.g., for potency, looking at orthogonal tests and IC50 values). There also should be another, independent control sample in those assays to compare with using the same dilution scheme to assess for drift of the reference standard.
The number and type of tests in a stability program should be justified, but selection of only stability-indicating attributes may be feasible. The strategy how to extend the period of use of the reference standard can be included in the initial protocol filed with the agency, with appropriate acceptance criteria. If those are approved, extensions can be recorded within an annual report or within the quality system.
How should reference standards be aliquoted and stored? Several attendees indicated that flash-freezing reference standard aliquots appear to be useful to maintain a homogenous, stable standard over time. Single-use aliquots used immediately and discarded after use are preferable. In such cases, stability data need cover only the long-term storage temperature and thawing step. However, if after thawing, aliquots are held at a different temperature then reused, in-use stability data need to be obtained. For example, if reference standards are thawed and held at 2–8 °C, then data should be collected to support a maximum hold time.
What is a typical time interval for reference-standard recalibration or stability exercises? An initial retest date for a frozen reference standard is typically one year. Most manufacturers carry out the stability time points annually unless/until sufficient data are collected to show long-term stability. However, that is risk-based and product dependent. To closely monitor degradation in early phase, some manufacturers test more frequently in the first year of use. If you have an unstable or new product, testing more often is recommended. If you have appropriate supporting stability data, then annual testing may be justified. A primary reference standard that you have on stability can be tested only once per year. You need to have enough replicates during a primary reference standard stability study for it to be meaningful for a given method (e.g., potency assay). Or you should at least justify how you could see a real shift with the number of assays carried out.
If a reference standard isn’t that stable and must be replaced often, how do you prevent drift or change in the standard? It could be justified to change the formulation of the protein standard specifically to create a stable reference standard as long as it can be shown that method performance is not adversely affected. If buffer components used for long-term stability of reference standards are different from those used with the drug substance and drug product (e.g., addition of stabilizers), then they should be shown not to affect the protein characteristics.
What is the value of testing for extractables and leachables from reference-standard containers? It is important to understand whether such compounds influence the assays (e.g., if new peaks appear) or change the protein itself over the lifetime of a reference standard. So we recommend performing such studies if there is a risk to the quality or accuracy of the stored reference standards.
What reference standard material should be used for comparability exercises? After a major manufacturing change, you may elect to use a primary reference standard as the comparator material. For minor changes, you could use the working standard, but that choice should be justified. Using a working reference standard appears to be more common in practice, depending on the level of characterization and origin of the working standard. Note that a reference standard is only one material used in a comprehensive comparability study; typically, several pre- and postchange batches are included to demonstrate normal process variability.
Part 2 of this article is available in our May 2014 archives.
Disclaimer
The content of this manuscript reflects discussions that occurred during the CMC Strategy Forum. This document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance documents, points-to consider documents, or direct discussions with the agency.
Author Details
Corresponding author Anthony Mire- Sluis is vice president, North America, Singapore, contract and product quality at Amgen Inc., amire@amgen.com. Nadine Ritter, PhD, is president and analytical advisor at Global Biotech Experts LLC, Barry Cherney is executive director of product quality at Amgen Inc., Dieter Schmalzing is senior principal advisor at Genentech, a Member of the Roche Group, and Markus Blümel is team leader, late-phase analytical development of biologics at Novartis Pharma AG.
REFERENCES