Mycoplasmas and related bacteria in the class Mollicutes are parasitic organisms found not only on the external surfaces of a wide range of eukaryotic host cells, but also intracellularly. They are characterized by small size and lack of a rigid cell wall, which gives them resistance to β-lactam antibiotics and the ability to pass through 0.2-µm filters (1,2). Contamination by Mollicutes is a common problem for cell cultures that is not easily detected because it usually does not produce turbidity or cytopathic effects even though high densities are achievable (105–108 organisms/mL) (3). A contaminated product-producing cell culture may not be apparent, but the contamination can affect cell growth and alter the metabolic and biochemical characteristics of contaminated cells — and it may lead to unsafe final products (2).
PRODUCT FOCUS: ALL BIOLOGICS
PROCESS FOCUS: PRODUCTION
WHO SHOULD READ: QA/QC, PROCESS DEVELOPMENT, PRODUCT DEVELOPMENT, AND ANALYTICAL PERSONNEL
KEYWORDS: LYOPHILIZATION, LIQUID NITROGEN, PARENTERAL PRODUCTS, BIOPHARMACEUTICALS
LEVEL: ADVANCED
Mycoplasma infection can originate from laboratory personnel, contaminated animal sera, and contaminated aerosols. It may be disseminated by the sharing of contaminated cells among different laboratories. The presence of mycoplasmas of swine origin in bovine serum may come from contamination in mixed slaughterhouses. Trypsin is a compound of swine origin often used in cell culture, and it can also contribute to the presence of mycoplasma DNA (4).
Mycoplasma species magnified DIAGNOSTIC MYCOPLASMA LABORATORY, UNIVERSITY OF ALABAMA AT BIRMINGHAM WWW.MYCOPLASMA.UAB.EDU
Five species of Mollicutes — Mycoplasma arginini, M. fermentans, M. hyorhinis, M. orale, and Acholeplasma laidlawii — account for ~95% of identified cell culture contamination (1,3). Testing is advised and/or regulated throughout the world for such contamination in master cell banks, working cell banks, virus seed lots, control cells, virus harvests, bulk vaccines, final batch lots, monoclonal antibodies, immunological modulators, interferon and other cytokines, growth factors, or any biological materials produced in cell substrates. In the United States, mycoplasma testing of cell lines and biological products derived from cells is regulated by Title 21 of the Code of Federal Regulations (21 CFR 610.30), the Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals document of 1993, and a 2006 draft guidance titled Characterization and Qualification of Cell Substrates and Other Biological Starting Materials Used in the Production of Viral Vaccines for the Prevention and Treatment of Infectious Diseases.
The biopharmaceutical industry is pushing for development of rapid mycoplasma testing as an alternative to standard 28-day cell culture methods. Nucleic acid amplification techniques (NATs) may be used as an alternative to the traditional method, as can an indicator cell culture method, if it can be demonstrated to be comparable with culture-based methods by suitable validation (5,6). In some cases, culture-based procedures cannot be used because of an inability to completely neutralize vaccine viruses, which necessitates the use of NAT-based assays to test for mycoplasma. Researchers began using the polymerase chain reaction (PCR) in the late 1980s to detect mycoplasma contamination in biological samples. Those early assays were limited to specific species, but today numerous broad–target-range assays have been developed (7,8,9,10). In addition, commercially available mycoplasma tests include both enzyme and NAT-based assays. But to our knowledge, there are none approved by the FDA to replace traditional cell-culture and broth methods for detecting mycoplasma.
Development of a NAT-based assay involves optimization of both the nucleic-acid extraction method and the PCR amplification reaction(s) to maximize assay sensitivity and specificity. Challenges include
-
targeting a large number of species within one class of bacteria without significant cross reaction with related bacteria or nucleic acids that may be present during biological product manufacture
-
evaluating fluorescent signals in samples when positive amplification indicates the presence of nucleic acids sequences but not necessarily the presence of viable mycoplasma.
Here we present an overview of a NAT-based detection assay that has been developed and validated for a wide range of Mollicutes including Mycoplasma and Acholeplasma based on guidelines in the European Pharmacopoeia (5). Our assay uses reverse transcription PCR (RT-PCR) with sequence-specific primers for amplification of target sequences and dual-labeled fluorescent probes for additional specificity. They were developed specifically for this assay to target mycoplasma 16S rRNA. Because RNA is typically more labile than DNA, its extraction and targeting (with an included DNase step) should decrease detection of nonviable organisms as well as persistent residual DNA. In addition, we expected the target RNA to be present in more copies per cell than its corresponding DNA, thus increasing the assay sensitivity.
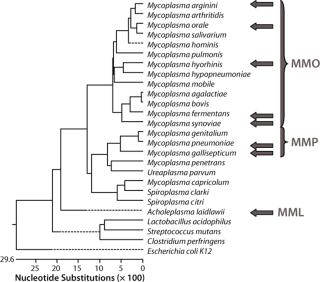
Materials and MethodsThis assay required a range that would include the Mollicutes but remain specific enough to discriminate between them and other bacteria. To maximize the range, sensitivity, and specificity of the assay, we tested three primer–probe sets in three reaction mixes based on the phylogenetic relationships within the Mollicutes class (1) using Lasergene software from DNASTAR, Inc. (www.dnastar.com). Master mix “Orale” (MMO) was used for the group of mycoplasma species that includes M. hominis, M. orale, M. arginini, M. fermentans, M. hyorhinis and M. synoviae. Master mix “Pneumoniae” (MMP) was used for the group that includes M. pneumoniae and M. gallisepticum. Master mix “Laidlawii” (MML) was used for the Acholeplasma species, notably A. laidlawii. In addition, a β-actin primer–probe set was used as an alternative target to detect the control RNA used in each extraction.
Validation included both quantitated nucleic acid targets and well-characterized Mycoplasma and Acholeplasma organisms as reference standards titered by determination of colony-forming units (CFU) on agar plates. To assess the assay at the 10-CFU/mL level, strains of M. arginini (ATCC #23243), M. fermentans (ATCC #19989), M. hyorhinis (ATCC #17981), M. orale (ATCC #23714), M. pneumoniae (ATCC #15531), and A. laidlawii (ATCC #23206) represented an optimal selection in terms of frequency of occurrence as contaminants in mammalian cell cultures and phylogenetic relationships. Growth studies involving seven-day broth cultures preceding RNA isolation used those strains as well as M. gallisepticum (ATCC #19610) and M. synoviae (ATCC #25204).
Both the RT-PCR method and traditional culture methods are performed routinely at our company, where they’ve been demonstrated to be comparable by their ability to detect 10 CFU/mL using the same titered stocks. To assess the molecular limit of detection (LOD) — the minimum number of molecules that can be routinely measured (>95% probability of generating a positive result) but not necessarily quantitated — we used plasmid copy number standards containing a portion of the 16S rRNA coding region from the appropriate organism. Designated as pAlaidlawii, pMpneumoniae, pMorale, pMfermentans, pMhyorhinis, and pMarginini, the plasmids were purified and quantitated by spectrophotometry. We tested multiple independent fivefold dilution series from 156,250 to two copies per reaction with a minimum of 24 replicates at each dilution.
An exogenous RNA control (total genomic RNA from a human cell line, HEK 293) was added to all samples before nucleic acid isolation and used for routine verification of the absence of inhibition as well as an overall control for the extraction, RT, PCR, and detection steps. We used additional plasmid positive controls to spike sample replicates with a defined copy number standard from representative target species to demonstrate achievement of the expected sensitivity. Specificity was also addressed using DNA isolated from bacterial species that have a close phylogenetic relationship to the Mollicutes class. Streptococcus mutans genomic DNA (ATCC #25175D), Lactobacillus acidophilus genomic DNA (ATCC #4357D), and Clostridium sporogenes genomic DNA (ATCC #3584D) were purchased from the American Type Culture Collection (www.atcc.org) and assayed at 250 pg/reaction (~100,000 genome equivalents).
We included five matrices in our validation: phosphate-buffered saline (PBS), mycoplasma broth (which includes yeast extract and horse serum), fetal bovine serum (FBS), cell culture supernatant derived from a murine hybridoma cell line (designated ERV514), and cell culture lysates derived from 107-cells/mL cultures of either ERV514 or Sp2/0 cells. These matrices were tested either unspiked or spiked with 10 CFU/mL of our panel of titered strains of mycoplasma.
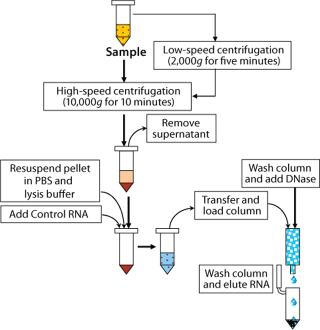
Extraction and purification of RNA from samples used a column purification system from Qiagen (www.qiagen. com). Samples were first lysed with a highly denaturing buffer containing guanidine thiocyanate. This buffer not only inactivated RNases, but also protected the isolated RNA from degradation. All samples included a control RNA, and we included an on-column DNase treatment to remove any contaminating DNA that may have been present, reducing the possibility of detecting free mycoplasma DNA or nonviable organisms (2). Only low-speed centrifugations were needed for debris removal from the cell lysates. All other samples were extracted following a high-speed centrifugation step to concentrate all organisms present.
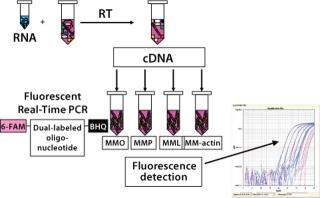
We performed a two-step RT-PCR on all samples and controls (3), with one reverse-transcription reaction per sample aliquotted into four PCR reactions. Those reactions were amplified using a sequence detection system from Applied Biosystems (www.appliedbiosytems.com), and the generated fluorescent signals analyzed by dedicated software. The results were expressed as CT (threshold cycle), the PCR cycle number at which a signal exceeded a threshold fluorescence value. All PCR reactions were performed in triplicate.
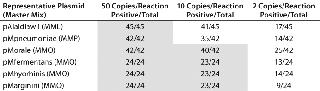
Molecular Limit of Detection, Linearity, Sensitivity, and Robustness: We tested multiple independent fivefold dilution series of the six plasmid copy number standards from 156,250 to two copies per reaction with a minimum of 24 replicates at each dilution (Table 1). All reactions above 10 copies generated positive signals. The demonstration of both positive and negative reactions at the two-copy level suggests that each plasmid target in each master mix has a one-copy sensitivity. Although some M. pneumoniae and A. laidlawii 10-copy reactions generated no positive signals, at least 33% of replicates did generate signals in all triplicate reactions. For the assay as a whole, using the suggested 95% positivity cut-off point for all reactions (5), the molecular LOD would be set at 50 copies.
Table 1: Molecular limit of detection (LOD) shows one-copy sensitivity for all copy-number standard plasmids. The LOD for the copy number plasmids is 10 copies/reaction using the MMO master mix and 50 copies/reaction for copy number plasmids using MML and MMP master mixes. All shaded boxes indicate a >95% LOD.
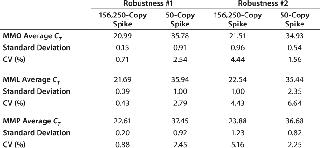
Standard curves were generated from the dilution series of the plasmid copy number standards and CT values: 65 out of 67 had slopes between -3.37 and -3.88, representing PCR efficiencies of 98.03–81.02%. One dilution series of pMpneumonia had a slope of -4.15 (74.17% efficiency), and one dilution series of pMorale had a slope of -4.05 (76.57% efficiency). All curves demonstrated linearity and correlation coefficients ≥0.9800. Our assay was shown to be robust by introducing 10% volume variations in the primer–probe and total reaction mixes, generating five different variations for each master mix. Neither the 156,250-copy spike nor a 50-copy spike was significantly affected by those changes, with coefficients of variation (CVs) of 0.71–4.44% for the MMO, 0.43–6.64% for the MML, and 0.88–5.16% for the MMP (Table 2).
Table 2: Robustness of the master mixes were tested by introducing 10% volume variations in the primer/probe and total reaction mixes. The coefficient of variation did not exceed 5.2% and 6.7% for the 156,250-copy and 50-copy spikes, respectively.
Specificity, Background Signals, and Cross-Reactivity: We evaluated specificity in several ways because a RT-PCR assay can detect any target nucleic acid in viable and nonviable organisms as well as residual nucleic acids from environmental sources. Reagent controls contained a minimal amount of biological materials, so we used them to assess the possible impact of residual mycoplasma nucleic acids. We included the PBS matrix for two reverse transcription control reactions: RT1 containing the control 293 RNA and RT2 without it. In the negative RT reactions and extraction controls, nine out of 372 (2.4%) reactions in MMO, 0/156 reactions in MMP, and 14/162 (8.6%) reactions in MML generated amplification signals equivalent to 2–11 copies, 0 copies, and 1–2 copies, respectively (Table 3).
Table 3: Target nucleic acid specificity is evaluated by analyzing the amplification observed in extraction and reverse-transcriptase reaction controls, typical mammalian and bacterial DNAs, and unspiked model matrices.
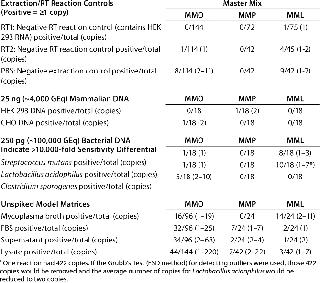
In 25 ng of mammalian DNA derived from HEK 293 or Chinese hamster ovary (CHO) cells, 1/36 (2.8%) reactions in MMO, 1/36 (2.8%) reactions in MMP, and 0/36 reactions in MML generated amplification signals equivalent to two copies, two copies, and 0 copies, respectively (Table 3). In 250 pg of bacterial DNA, 7/54 (13%) reactions in MMO, 0/54 reactions in MMP, and 18/36 (50%) reactions in MML generated amplification signals equivalent to 1–10 copies, 0 copies, and 1–7 copies, respectively (Table 3). In all master mixes the mycoplasma broth (containing horse serum), FBS, cell culture supernatant, and cell lysate had sporadic amplification signals: 126/432 (29%) reactions in MMO, 10/114 (14%) reactions in MMP, and 20/114 (18%) reactions in MML generated amplification signals equivalent to 1–220 copies, 1–22 copies, and 1–11 copies, respectively (Table 3).
The calculated copy number values derived from reagents alone, (HEK 293 RNA, PBS, mammalian DNA, and bacterial DNA) suggested that these signals were not caused by cross-reactivity of primers and probes with the sample DNA, but rather indicated residual mycoplasma-derived nucleic acids. Even if the signals in bacterial DNA resulted from cross-reactivity, it would indicate a >10,000-fold differential in sensitivity between these species and the target Mycoplasma organisms. These experiments were performed in an ongoing testing laboratory, so the FBS, supernatant, and lysate were processed in an environment that isolated those potential sources from materials and samples known or expected to be free of adventitious agents. That required some exposure to potential sources of Mycoplasma or related nucleic acids.
To investigate whether the signals generated in the five matrices indicated actual viable mycoplasma contamination, we subjected them to traditional detection methods including a cell culture broth method, an agar method, and an indicator cell/DNA staining method (Tables 456). All matrices used in this study (including two lots of mycoplasma broth) that may have generated amplification signals using the mycoplasma-specific RT-PCR were shown to have no viable Mycoplasma by the three methods. This supports the conclusion that the positive signals indicated the presence of nucleic acid sequences and not viable Mycoplasma. These results highlight the fact that routine testing of biological materials may generate small mycoplasma-specific amplification signals.
Table 4: Confirmation of nonviability by broth method (two independent assays performed in duplicate); no viable Mycoplasma was detected in any matrix or negative control (shaded boxes).
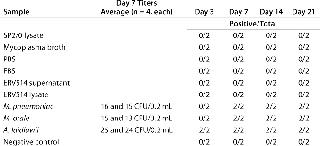
Table 5.Confirmation of nonviability by agar method (two independent assays performed in quadruplicate); no viable Mycoplasma was detected in any matrix or negative control (shaded boxes).
CFU Limit of Detection: Our major goal in the development and validation of this assay was detection of 10 CFU/mL in multiple matrices with a broad range of Mollicute species. So before the initial centrifugation steps in RNA extraction, the five model matrices were spiked to a final concentration of 10 CFU/mL with M. arginini, M. fermentans, M. hyorhinis, M. orale, M. pneumoniae, and A. laidlawii. Following the RT-PCR reactions, we used the resulting fluorescent amplification signals to calculate a copy number value based on the plasmid copy number controls (4). This calculated value represents 25% of the total cDNA copies from the RT reactions. The 10 CFU/mL target of sensitivity for this assay was achieved in 100% of reactions in four matrices tested.
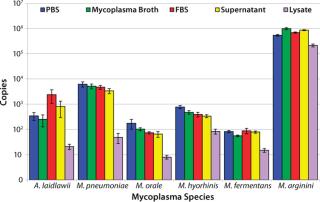
Nearly all of the RT-PCR reactions (714/720 or 99.2%) using the spiked samples were positive for amplification, with the limited number of failed reactions found only in the lysate matrix. The lysate had been subjected to a low-speed spin to remove cell debris before the highspeed spin that concentrates potential mycoplasma organisms. Although that low-speed spin helped process the dense lysate for RNA extraction, it may have removed a portion of the organisms present due to mycoplasma–cell-membrane binding.
Because mammalian cell banks are routinely tested for the presence of Mycoplasma, it is important that the concentration of cells in any lysate be optimized to improve the process efficiency. Future testing will determine the maximum number of cells that can be processed efficiently using this extraction method.
The lowest average mycoplasma cDNA copy number detected from A. laidlawii 10-CFU/mL spikes in PBS, mycoplasma broth, FBS, and cell culture supernatant tested in MML was 253±126, whereas the lowest average mycoplasma cDNA copy number detected from the M. pneumoniae 10-CFU/mL spike in the same matrices tested in MMP was 3,401±767. For M. arginini, M. fermentans, M. hyorhinis, and M. orale, the average mycoplasma cDNA copies detected from a 10-CFU/mL spike ranged from 56±4 (M. fermentans in mycoplasma broth) to 1,000,855±95,180 (M. arginini in mycoplasma broth) (4). This roughly four-log range in cDNA copies for the 10-CFU/mL spikes from different species reflects the highly variable nature of the number of mycoplasma genomes found in a CFU.
Alternate Target Reactions: All alternate target reactions using a primer–probe set for β-actin produced positive amplification signals. The successful β-actin reactions included extractions with all five matrices and RT1 reactions. We evaluated the ▵CT method to determine whether extraction efficiency could be quantitated based on RNA amplification from each column extraction relative to that in the RT control (RT1). If extraction efficiency was 100% for samples spiked with HEK 293 RNA, the same amount would be assayed in each alternate target reaction. By evaluating the difference in the CT values from a sample and RT1 in the formula, we could express 2CT the amplification from each sample as a fold difference. The larger was the number for a sample, the larger was the related fold difference compared with RT1, which suggests either a <100% extraction or some inhibitory effect of the matrix.
We observed trends for larger 2CT values in β-actin reactions associated with smaller copy numbers detected in CFU spikes in certain matrices, especially the lysate (data not shown). Unfortunately, there was no absolute correlation between the two values. Again, this supports the use of HEK 293 RNA as an extraction control but not as a quantitative indicator.
Intermediate Precision, Repeatability, and Ruggedness: Nearly all (99.8%) CVs of triplicate CT values (intraassay) for copy-number standard values between 156,250 and 50 in the 67 assays performed (1,206 reactions) fell below 3%. And nearly all (99.6%) CVs of triplicate CT values in the 48 assays performed with CFU spikes (720 reactions) fell below 4%. CVs of triplicate threshold cycle values for β-actin reactions predominantly fell below 2%.
Three analysts performed these experiments over several months using two sequence detection systems. The interassay precision was evaluated by CV of CT values and ranged 0.39–5.42% for all copy number standard values with all six target species between 156,250 and 50 copies in the 67 assays performed with the plasmid copy number standards.
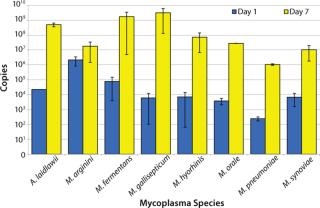
Pre-extraction Growth Enrichment Analysis: Because a fraction of the samples tested in this validation generated small amplification signals, we wanted to explore addition of a pre-extraction growth step. We expected this extra step to provide an enrichment of low-level viable mycoplasmas and an initial dilution of the original sample, thus diluting any potential inhibitors to RT-PCR and delineating viable and nonviable sources of mycoplasma nucleic acids.
A. laidlawii, M. arginini, M. fermentans, M. gallisepticum, M. hyorhinis, M. orale, M. pneumoniae, and M. synoviae were each inoculated into mycoplasma broth at a concentration of 1 CFU/mL. Aliquots were removed at days 0, 1, 2, 3, 4, and 7, then the material was subjected to the same RNA extraction and RT-PCR as described above. This preliminary study was performed twice in triplicate and revealed a significant increase in the number of cDNA copies (minimum 3 logs) for all species tested except for M. arginini (5). That exception most likely comes from a ratio of high genome copy to CFU. Because the number of M. arginini organisms in a broth culture is greater than expected based on the CFU titer, the potential for additional growth was limited. Future studies with M. arginini should include dilutions of the stocks to <1 CFU/mL for better understanding of its growth potential.
We developed a NAT-based detection assay and validated it for a wide range of Mollicutes including Mycoplasma and Acholeplasma using RT-PCR with sequence-specific primers and fluorescent probes. The assay has been validated based on the 2.6.7 guidelines of the European Pharmacopoeia using several matrices spiked with A. laidlawii, M. fermentans, M. hyorhinis, M. orale, M. pneumoniae and M. arginini. This panel provided an optimal selection in terms of cell culture contaminant frequency and phylogenetic relationships, and the assay achieved the suggested 10-CFU/mL sensitivity for comparability with culture-based methods. The negative control samples yielded detectable amplification fluorescence in 4–9% of sample reactions; however, the calculated copy number values of target sequence in these reactions were 1–2 copies per reaction. In addition, we subjected samples that tested positive to traditional culture methods (and time courses), and viable mycoplasmas were neither observed nor detected in any of those samples. The results of these studies indicated that this mycoplasma detection procedure is sensitive, specific, linear over appropriate ranges, and demonstrated acceptable precision, repeatability, ruggedness, and robustness.