Adenoassociated virus (AAV) vectors are a popular choice for modern gene therapies because of their favorable safety profile, low immunogenicity, and the ease with which they can be transduced into different cell and tissue types. An AAV genome is a single strand of DNA comprising a replication (rep) gene, which encodes regulatory proteins involved in genome replication, and a capsid (cap) gene, which produces three capsid proteins.
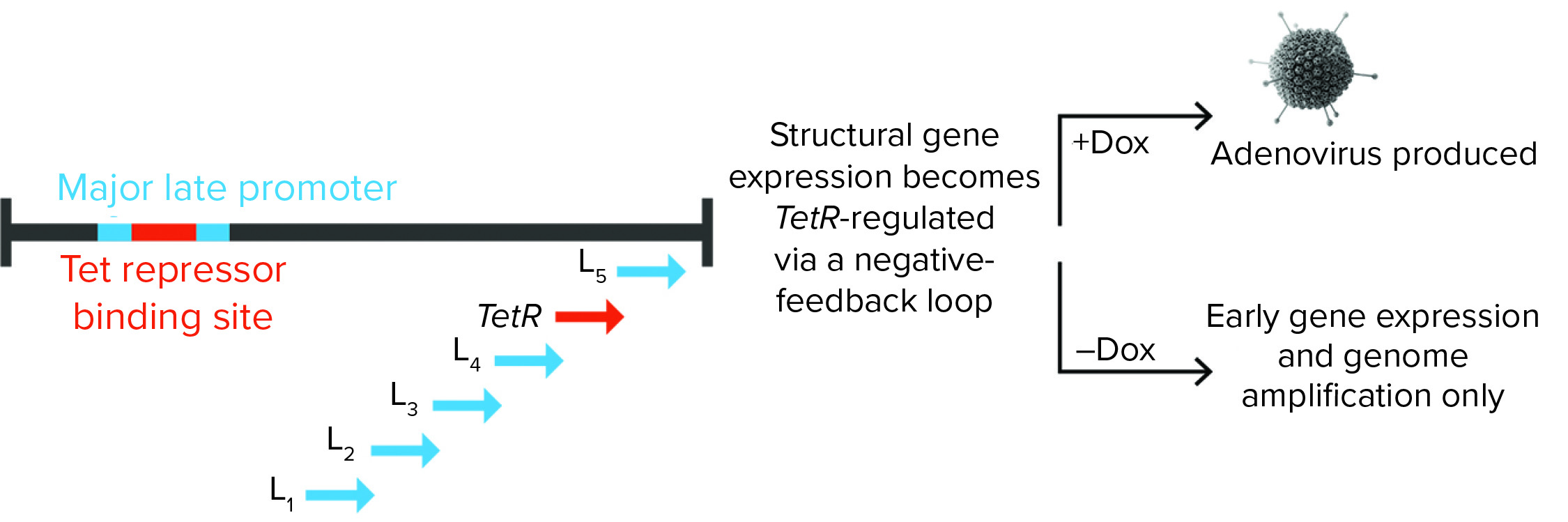
Figure 1: Tet repressor (TetR) binding site is inserted into the major late promoter, and the TetR gene itself is encoded within the late region. That creates a negative feedback loop, in which adenoviral structural proteins can be produced only in the presence of doxycycline (Dox). Without it, TetR binding inhibits expression of adenoviral late-region (structural) genes.
However, AAVs cannot replicate alone. In nature, AAV shares an exquisite relationship with adenovirus, which provides most of the functions needed for the AAV lifecycle. That highly evolved and dynamic relationship results in exceptionally high-fidelity production of AAVs, with almost every AAV particle receiving the correct genome (1).
As such, “helper” adenoviruses were important tools for manufacturing recombinant AAV vectors for early gene therapies. For such products, rep and cap genes are replaced in an AAV genome with a gene of interest. However, using wild-type adenovirus for AAV production means that the resulting viral vector preparation frequently contains as many helper adenoviruses as it does AAVs. That is a significant safety issue, and it makes the downstream removal of helper adenoviruses complicated and costly.
Over the past decade, three-plasmid transfection into recipient cells has become the predominant method of manufacturing AAVs. In this system, the first plasmid contains the rep and cap genes. The second plasmid contains a gene of interest flanked by the AAV genome inverted terminal repeats (ITRs). The third plasmid contains adenoviral “helper” genes.
This helper-free (or adenovirus-free) method of AAV production provides a safe and clean manufacturing approach, but it is not without its drawbacks. Scalability is the primary challenge and is in fact a critical issue facing the entire gene therapy industry because of the need to manufacture large quantities of AAV for clinical production of gene therapies.
A New Manufacturing Approach
For the past three years, a team at Oxgene has had one mission: to fundamentally change the way AAVs are manufactured. The team started with the conceptual premise that replicating what occurs in nature — using adenoviruses to make AAVs —would be the best approach. Thus, the team set out to investigate whether it could engineer adenoviruses such that they would generate high AAV yields without concomitant adenovirus contamination.
The team took advantage of the fact that adenoviruses have both early and late intracellular lifecycle phases. The early phase is crucial for AAV production. But the late phase, which is responsible for making adenoviral structural proteins, is not required at all. So, by regulating the late phase of an adenoviral lifecycle and switching the process off during AAV production, the team could use adenoviruses as helpers for efficient AAV manufacturing without adenovirus contamination. When more adenoviral vectors are needed, late gene expression simply is turned back on, and the system produces high titers of adenovirus.
To ensure supremely tight regulation of the adenovirus late phase, the team built in a proprietary negative feedback loop. That was achieved by placing a Tet repressor (TetR) binding site into the major late promoter (MLP), which drives late region gene expression, while the TetR gene itself is encoded within the late region (Figure 1). The team demonstrated that this process can repress adenovirus production by 99.999–100% during an AAV manufacturing run, and it can increase rAAV yield compared with either Ad5 helper plasmid or wild-type adenovirus.
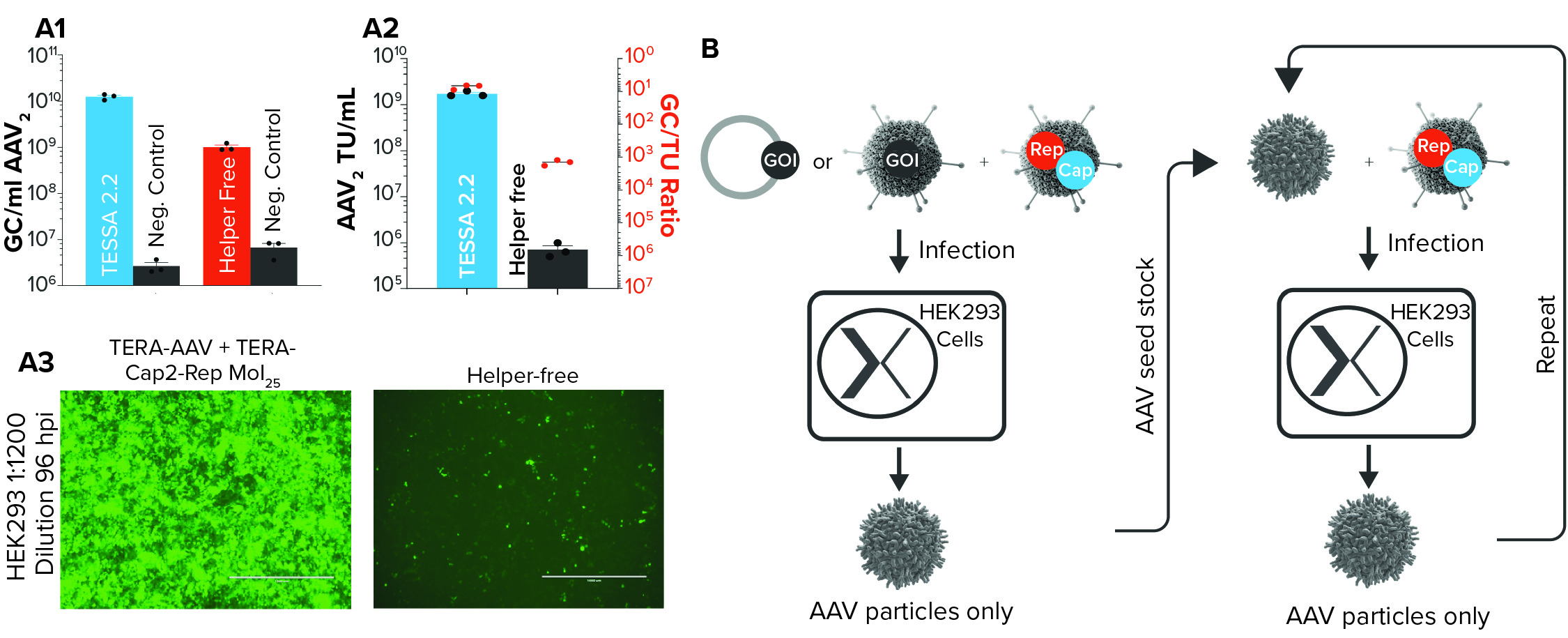
Figure 2: (a1) AAV2 genome copies derived from either Tet-enabled self-silencing adenoviral (TESSA) 2.2–based manufacturing (in which two adenoviral vectors deliver all AAV2 components to a cell) and a plasmid-based manufacture (helper-free) approach; negative controls do not include Rep and Cap in the respective process, and N = 3. (a2) Infectious AAV2 units per milliliter when manufacturing using either the TESSA system or plasmid-based process and the relative ratio of AAV2 genome copies to transducing units (TU) in HEK293 cells; (a3) images of HEK293 cells infected with filtered lysate of AAV2, produced using either a TESSA system or plasmid-based process; fluorescence indicates particle infectivity. (b) Schematic diagram of TESSA-based AAV manufacturing process
Scalability
Although the above results were positive, they still did not address the problem of scalability because the process still required plasmid transfection. The question was, Could rep and cap genes be inserted into engineered adenovirus to reduce the number of plasmids required for AAV production? The answer was that yes, they could. By doing that, the team produced stable adenoviral vectors that contain both rep and AAV cap genes. This result was the first time both genes have been included stably within an adenoviral vector. That enabled the efficient, simultaneous delivery of all adenovirus helper functions, alongside all AAV components, in a single agent without transfection. The process has been demonstrated for rAAV2, rAAV9, and rAAV6.
The only outstanding component required for full AAV production is the AAV genome encoding the gene of interest. That can be delivered by plasmid transfection, a second engineered adenoviral vector (which gives more efficient delivery than transfection), or a cell line with an integrated AAV genome. Oxgene researchers tested all three options, and the use of a second engineered adenoviral vector provided the best results. For rAAV2, the process achieved a 20-fold increase in AAV particle yields compared with the helper-free system. We also saw a 2,420-fold cumulative increase in AAV2 infectious yield (Figure 2a).
With those results, the potential of the new manufacturing system is apparent. The research team realized that once it had made and harvested the first AAV preparation, researchers could infect cells with both that AAV and more of the engineered adenoviral vector, simultaneously. Doing so would propagate the AAV seed stock further, producing more pure AAV in a simple, easily scalable process that was free of contaminants, plasmids, chemicals, and transfections (Figure 2b).
Oxgene’s technology represents a major paradigm shift in AAV manufacturing. It is completely scalable and significantly reduces the number of input materials required. If you already have good manufacturing practice (GMP)-grade AAV, you simply can grow more of it. If you only have a plasmid with your AAV genome in it, then you can recover a small amount of AAV and then grow it repeatedly, without ever needing more plasmid.
Oxgene anticipates that its process will forever change the way AAV is manufactured, thus reducing consumer costs of medications, improving product quality, and making safe gene therapies available to those in need.
References
1 Zeltner N, et al. Near-Perfect Infectivity of Wild-Type AAV As Benchmark for Infectivity of Recombinant AAV Vectors. Gene Ther. 17, 2010: 872–879; https://doi.org/10.1038/gt.2010.27.
Ryan Cawood is chief executive and founder of Oxgene, Medawar Centre, Robert Robinson Avenue, Oxford, UK; 44-1865-415-107; business@oxgene.com; www.oxgene.com.