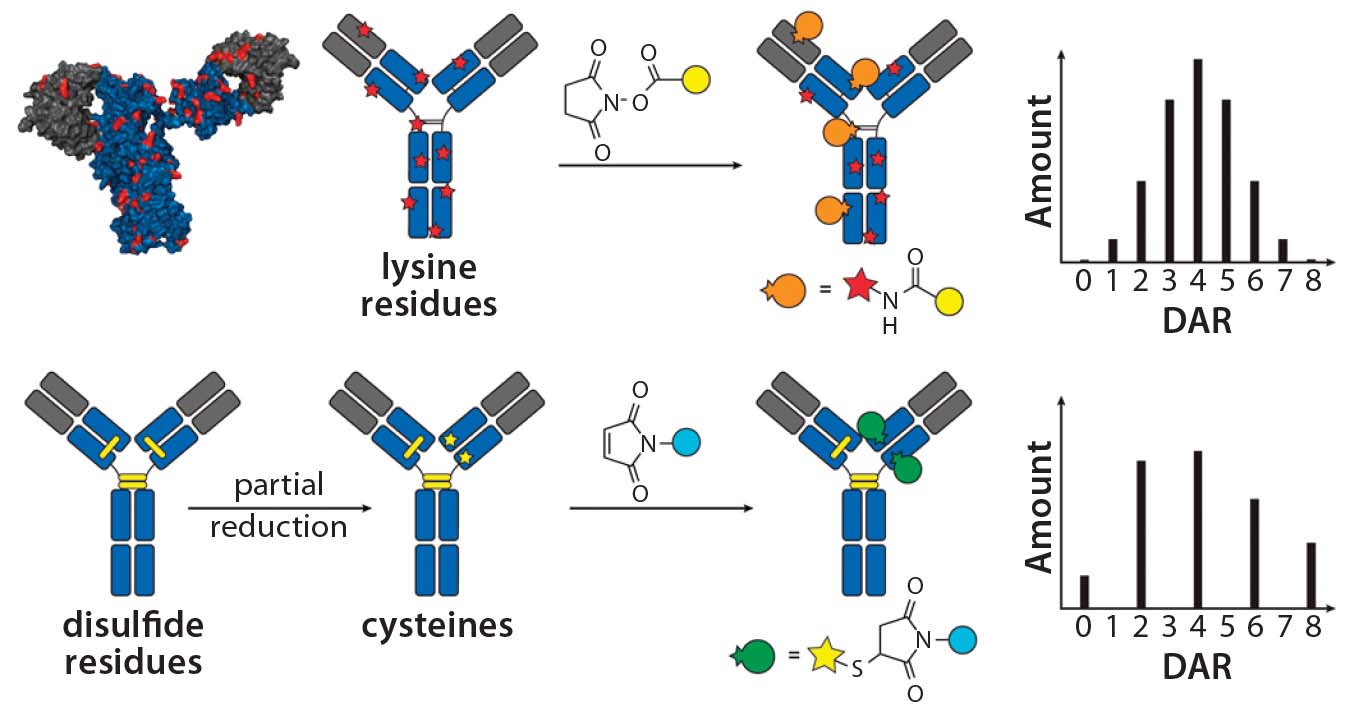
Figure 1: Schematic of lysine and cysteine conjugates and their expected DAR profiles.
Bringing a new biologic drug to market is a long and expensive process, with research and development (R&D) cycles that can span up to 15 years and may cost over a billion dollars. Biologic drug development also involves significantly more complex manufacturing and CMC components than does development of small molecules. Nonetheless, the pharmaceutical industry is increasingly shifting its R&D efforts to focus on biologic drugs. According to a recent report from Tufts Center for Study of Drug Development, the number of clinical trials involving biologic drugs has risen by over 155% from 355 in 2001 to more than 900 in 2012 (1). The same report reveals that biologics accounted for 71% of revenue from the world’s top-10 selling drugs in 2012 compared with a mere 7% in 2001. Worldwide sales of biologic drugs increased from US$36 billion to $163 billion during the same period.
Biologic drugs represent tremendous investment value for the pharmaceutical industry. They provide highly differentiated therapeutic options for difficult-to-treat diseases and thus can command higher prices while remaining less susceptible to cheaper generic competition than classical drugs are. The top three product categories in biologics are monoclonal antibodies (MAbs), vaccines, and recombinant proteins — accounting for 37%, 27%, and 10% (respectively) of the total biologic medicines in development. Bioconjugation has been implemented in successful drugs from each of those product classes (2).
Bioconjugation strategies involve covalently linking a protein or peptide (biologic) with a small molecule, carbohydrate, oligonucleotide, synthetic polymer, or another protein/ peptide. This approach can be crucial to create differentiation and drive product development in the highly competitive biologics market. These strategies were fundamental to development of highly successful conjugate vaccines such as Prevnar 13, Menactra, Menomune, and HibTITER. Those four were created by conjugating bacterial polysaccharides to immunogenic carrier proteins (3). Similarly, bioconjugation to half-life–extending polymeric carriers such as polyethylene glycol (PEG) is applied to create drugs — e.g., certozilumab (Cimzia), pegfilgrastim (Neulasta), and pegvisomant (Somavert) — that have longer duration of action than their unconjugated counterparts and dosing regimens that facilitate patient compliance (4).
Bioconjugation is the bridge that enables a combination of precise targeting and long half-lives of MAbs with the cytotoxic killing power of high-potency toxins to create a new class of drugs called antibody–drug conjugates (ADCs). Two recent ADC approvals — brentuximab vedotin (Adcetris) for Hodgkin’s and anaplastic large-cell lymphomas and ado-trastuzumab emtansine (Kadcyla) for HER2- positive breast cancer — have provided validation for the ADC concept and fueled intensive R&D in all aspects of ADC research (5).
ADCs have breathed new life into the oncology-targeted MAb market. Presently, nearly 40 different ADCs are in clinical development for treatment of both solid and blood-borne cancers (6). The choice of conjugation chemistry is key to efficacy, disposition, and toxicity of ADCs, which has spurred development of next-generation bioconjugation methods. Herein we highlight significant developments in bioconjugation methods that can drive creation and selection of optimal ADC molecules while aiding manufacturing and characterization of these complex bioconjugates.
First-Generation ADCs
An ADC consists of a MAb connected to a cytotoxic payload by means of a chemical linker. The combination of those two historically successful therapeutic classes of molecules should provide a drug that is greater than the sum of its parts. From an antibody perspective, combination with a cytotoxic agent can significantly enhance potency and tumor-killing potential (increased efficacy). From a cytotoxin perspective, combination with a targeted antibody can increase selective delivery to tumor cells while minimizing exposure to normal, healthy tissue (which increases safety and tolerability).
The choice of linkers and conjugation chemistry for making first-generation ADCs was dictated by limitations of working with proteins. Thus, linkers were functionalized with reactive groups designed to specifically react with surface-accessible nucleophilic amino-acid side chains belonging to native amino acids such as cysteine (thiol) or lysine (amine) (Figure 1).
Lysine Technology: Linkers bearing active esters — e.g., N-hydroxysuccinimide or sulfosuccinimide — readily react with solvent-accessible lysine residues on an antibody backbone. However, because ~80 reactive lysines are available for chemical elaboration, precise control of the conjugation site is not possible with that technology. On average, lysine-directed conjugations result in heterogeneous mixtures containing ADC molecules bearing as few as zero and as many as eight drug molecules per antibody. Peptide-mapping experiments have revealed that conjugates can be detected with attachment occuring at ≤40 lysine residues spanning both the heavy and light chains (7). Nonetheless, this method has proven useful in creation of the Kadcyla product mentioned above along with a number of other ADCs currently in different stages of development.
Cysteine Technology: Thiols in native cysteine residues are usually tied up in pairs of disulfide bonds and not available for conjugation. However, treatment of an antibody with reducing agents such as dithiothreitol (DTT) or tris(2- carboxyethyl) phosphine (TCEP) can break those disulfide bonds and expose free thiols, which then can be readily conjugated with maleimide-containing linkers (Figure 1) (8). Up to four interchain disulfide bonds can be reduced, thereby exposing up to eight reactive thiol groups for conjugation. Conditions developed for thiol chemical conjugation lead to either complete or partial reduction of disulfide bonds, and conjugates made using this method can contain either zero, two, four, six, or eight drugs per antibody molecule (9).
It is important to note that beyond the number of drugs per antibody molecule, another level of heterogeneity comes from the site of conjugation. Thus, an ADC with a specific drug-to-antibody ratio (DAR) generated by cysteine conjugation is still a heterogeneous mixture of conjugates with different sites of conjugation. However, it is fair to say that because only eight sites are available for cysteine conjugation (compared with up to 80 available for lysine-directed chemistries), the cysteine conjugation approach provides greater control over the site of conjugation, facilitating better characterization. The controlled reduction–alkylation strategy has been used successfully for making the approved Adcetris product along with a number of other ADCs currently undergoing clinical trials (10).
Stoichiometry: In 2004, researchers at Seattle Genetics studied the in vivo effects of ADCs targeting CD30+ tumor cells — with two, four, and eight monomethyl auristatin E (MMAE) toxins per antibody molecule — and demonstrated for the first time that the stoichiometry of drug loading significantly influenced the drug’s pharmacokinetics (PKs), efficacy, and toxicity (11). Hamblett et al. found that in their system, ADCs with four drugs per antibody were more potent than those with two but had comparable efficacy and better tolerability than those with eight drugs/antibody. The results indicated that, in general, ADCs with higher drug loadings have greater clearance, more efficacy, and increased toxicity. That implied that each ADC would have an optimal drug loading with the right balance of efficacy and toxicity.
That seminal work established the concept that the DAR is a key design parameter for ADCs. It became apparent that chemical conjugations to native cysteine or lysine residues would be suboptimal because they produce heterogeneous ADC mixtures. Heterogeneity comes from differences in DAR and conjugation sites, resulting in ADC subpopulations that may be less potent, more toxic, and have differing disposition and PK properties. In addition, analytical characterization and controlling batch-to-batch variability during manufacturing remains a significant challenge with such nonselective conjugation methods. To overcome those limitations, the concept of site-specific conjugations has evolved, initially with a goal of producing homogeneous ADCs and controlling DAR and sites of conjugation.
The Quest for Homogeneous Products
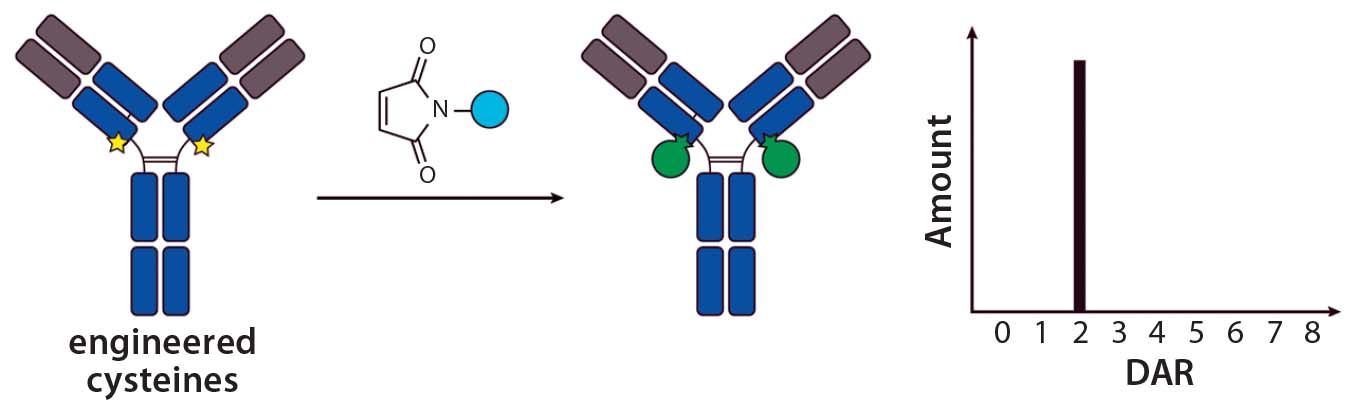
Figure 2: Site-specific THIOMAB conjugation and its expected DAR profile
Site-Specific Conjugation with THIOMABs — a New Era in ADC Conjugation: The pioneering work describing the first site-specific conjugation was reported by Genentech, who together developed methods to engineer reactive cysteine residues at specific positions on an antibody and use them for conjugation without affecting the native interchain disulfide bonds (12). Antibodies with those reactive cysteine residues were referred to as THIOMABs, so their ADCs were called THIOMAB–drug conjugates (TDCs) (Figure 2).
Jununtula et al. compared the properties of an ADC directed against ovarian cancer antigen MUC16 prepared by conventional cysteine conjugation to MMAE with a TDC using the same antibody– payload combination (12). They reported that, even though the TDC carried half the amount of cytotoxic payload, it was as potent and efficacious as the ADC in both in vitro and in vivo models. An impressive finding was that the TDC was much better tolerated by both rats and cynomolgus monkeys than was the conventional ADC, which demonstrates that an improved therapeutic index was achieved with the site-specific conjugation method.
The same team has also reported that a TDC version shows comparable efficacy and improved safety over trastuzumab emtansine, an ADC made using conventional lysine conjugation of a HER2-targeting antibody with a maytansine payload (13). As reported in 2013, an industry– academic collaboration led by Seattle Genetics incorporated engineered cysteine residues in an anti-CD70 MAb heavy chain and used it to conjugate extremely hydrophobic payloads. The resulting ADCs had uniform drug loading, lower aggregation, and an overall superior profile compared with those made using conventional cysteine conjugations in the reduced hinge regions (14).
Continued work with THIOMABs at Genentech has revealed another fundamental concept: Not only is ADC homogeneity key to improved biophysical and therapeutic properties, but the actual site of conjugation on the antibody backbone also has a major influence on in vivo behavior of an ADC molecule. Shen, et al., made multiple homogeneous TDC conjugates with a HER2-targeting antibody using a MMAE payload, wherein the engineered cysteine for conjugation was located in either the light chain (LC), heavy chain (HC), or Fc region of the antibody (15). All conjugates demonstrated comparable in vitro potencies, but the authors report significant differences in their in vivo efficacy and PK properties. The LC conjugate demonstrated the greatest efficacy when studied in a mouse xenograft model, in which the HC conjugate had moderate and the Fc conjugate had little to no activity. A mouse PK study revealed a similar trend with the LC conjugate demonstrating the greatest stability and lowest clearance followed by the HC conjugate, and the Fc-conjugated ADC was cleared fastest and provided the lowest ADC exposure (15). These results were attributed to differences in the local microenvironment and solvent accessibility contributing to differential stability of the linker system at different sites.

Table 1: Site-specific, next-generation conjugation technologies
More Than Just Homogeneous ADCs
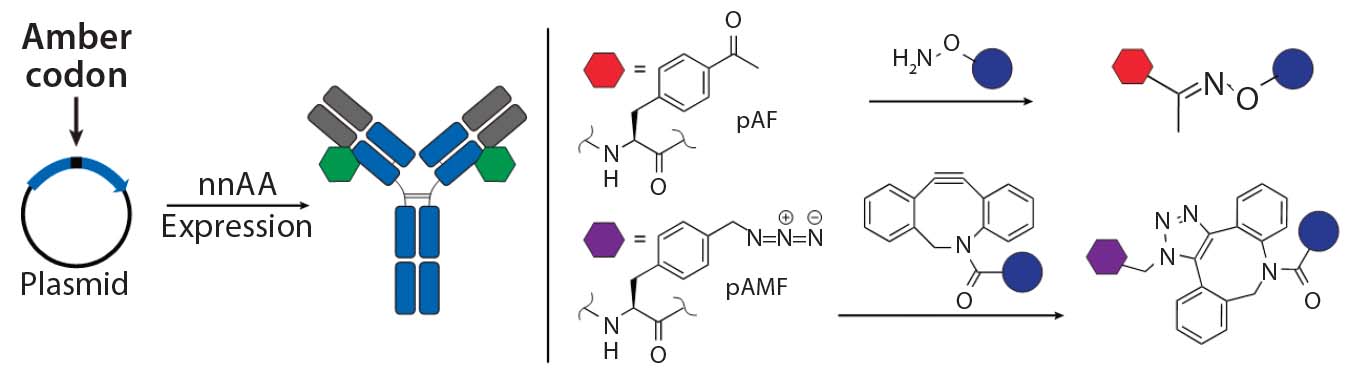
Figure 3a: Site-specific bioconjugation approach for generating ADCs with unnatural amino-acid
incorporation
Early THIOMAB studies served as a catalyst for developing several promising site-specific bioconjugation methods in the quest for making homogeneous ADCs (Table 1, Figure 3). All these methods can deliver homogeneous ADCs relative to the first-generation lysine and cysteine conjugations, but only a subset of these technologies offer greater versatility in finding the optimal conjugation site for a given antibody– payload combination. Those include
-
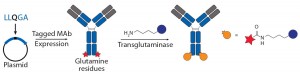
Figure 3b: Site-specific bioconjugation approach for generating ADCs through chemoenzymatic
bioconjugation using transglutaminaseGenentech’s THIOMAB technology
- conjugation by nonnatural amino acids (nnAAs) introduced by genetic-code modification at Ambrx, Sutro Biopharma, and Allozyne (16, 17)
- transglutaminase (TG) mediated conjugations to engineered glutamine tags by Pfizer’s Rinat group (18)
- conjugations with aldehyde-tagged antibodies generated by a coexpressed formylglycine-generating enzyme (FGE) at Redwood Bioscience (19).
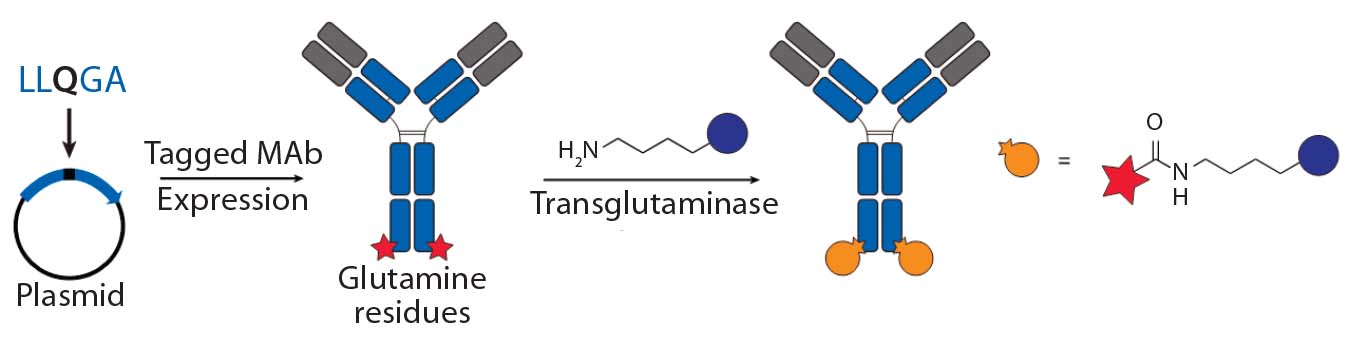
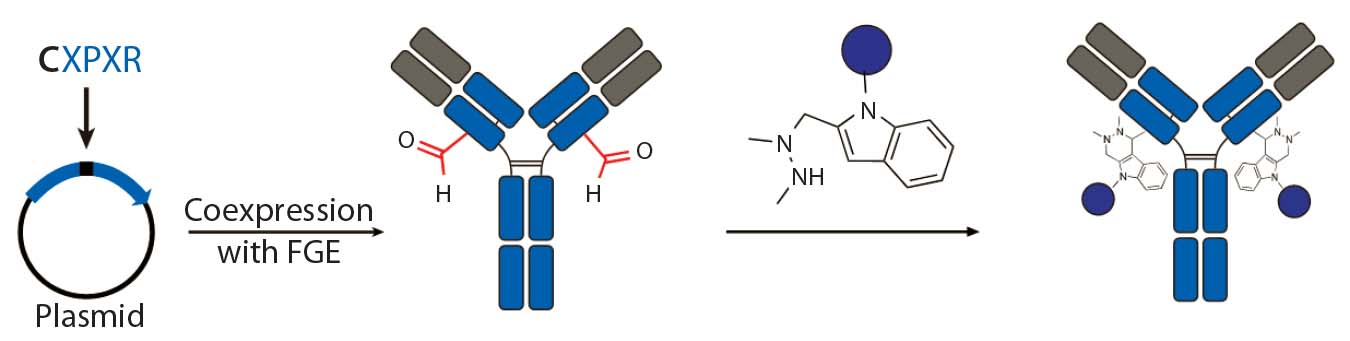
Figure 3c: Site-specific bioconjugation approach for generating ADCs through chemoenzymatic
bioconjugation using formylglycine-generating enzyme (FGE)
Those methods take advantage of protein engineering to move conjugation sites across different regions of an antibody and enable a true exploration of how conjugation sites determine the properties of ADCs. Recent studies at multiple laboratories, using distinct ADCs and different site-specific conjugation methods, give more credence to the idea that the site of conjugation determines the in vivo performance of an ADC molecule. Because such methods rely on protein engineering, they readily allow creation of homogeneous ADCs with DARs >2 when desired.
nnAA Incorporation: Several groups have developed recombinant protein expression systems with designed transfer ribonucleic acid synthetase pairs (tRNA–tRNA) that can incorporate nnAAs with bioorthogonal reactive “handles” onto any desired location on an antibody backbone. The reactive handles on such nnAAs serve as sites of conjugation to create site-specific ADCs (Figure 3a). Working with academic collaborators, Ambrx has
successfully incorporated p-acetyl phenyl alanine (pAF) in multiple antibodies using mammalian expression systems, then used the aryl ketone side chain of pAF to conjugate toxins via oxime bonds (16). Ambrx and Agensys have shown with multiple examples that ADCs with pAF NNA generally have comparable efficacy but improved PK and safety than either engineered or conventional cysteine-conjugated ADCs (20, 21). The authors attribute that to improved stability of the aryl oximes relative to maleimide thioethers in cysteine ADCs.
Tian et al. showed that the site of conjugation can have a dramatic effect on stability of NNA ADCs with protease-cleavable linkers (20). They found that in a 72-hour stability study using rat plasma, the ADC sheds about six times more payload when conjugated at position 115 in the heavy chain than when the conjugation is just one amino acid away at position 114. Sutro Biopharma reported incorporation of nnAA with an azide side chain into antibodies produced in a cell-free expression system (17). The azide handle was used to conjugate toxin payloads using either copper-catalyzed or copper-free “click” chemistry. Expression within a cell-free expression system can facilitate rapid exploration and characterization of various sites. Zimmerman et al. reported scanning more than 200 sites to find the optimal sites (offering the best conjugation efficiency and biological properties) for incorporation of nnAA (17). Rates of conjugation and stability of the triazole adducts are reportedly superior to oxime conjugates (22).
Transglutaminase Conjugations: Transglutaminase (TG) is an enzyme that catalyzes amide bond formation between a glutamine (Q) amino-acid side chain (acyl donor) and a primary amine (acyl acceptor). It has been used for making ADCs through conjugation of specific glutamine side chains with amine-functionalized linkers and payloads (Figure 3b) (23). Jeger and Shibli showed that TG does not modify natural glutamine residues in an antibody backbone but that, in deglycosylated antibodies, Q295 can serve as a substrate for TG for conjugation (24). Two research groups have used that observation to create site-specific ADCs through TG-mediated conjugation at natural Q295 or engineered N297Q mutants in the Fc region of antibodies (24–26).
Those TG conjugation methods limit the site of conjugation to the fixed residues. But Strop, et al., identified a short glutamine-containing tag (LLQG) that served as a TG substrate, then incorporated it into 90 surface-accessible locations across an antibody backbone and tested those sites for conjugation with an amine-linker–functionalized payload (26). This Rinat group identified 12 sites distributed across different antibody domains that showed efficient conjugation, and they were able to make ADCs with both cleavable and noncleavable linkers and with diverse payloads. The researchers studied the effects of conjugation site by comparing ADCs with two distinct sites, one with heavy-chain (HC) conjugation and one with a light-chain (LC) conjugation site. The authors reported that both those ADCs were comparable in efficacy but better in safety and tolerability than a conventional ADC. As with the nnAA and THIOMAB conjugates, TG conjugation also demonstrated that the site of drug loading can significantly affect antibody PKs and linker stability because ADCs with conjugation at HC seemed to have faster clearance. That effect became more pronounced in rats than in mice. The TG conjugation process is scalable and compatible with multiple payloads, it forms generally stable amide bonds in conjugates, and it requires no nnAA incorporation.
Aldehyde-Tagged Antibodies (SMARTag Technology): Redwood Bioscience has pioneered a novel chemoenzymatic approach that uses the naturally occurring formylglycine-generating enzyme (FGE) to introduce a formyl glycine (fGly) residue onto protein backbones that serves as a handle for site-specific conjugation (19). This method enables us to undertake a structure–activity relationship (SAR) study at the conjugate level and design site-specific ADCs with excellent potency and improved safety (27). Unlike the transglutaminase approach, the enzyme is used to create the conjugation handle in this case rather than catalyze the conjugation process. As a result, that chemoenzymatic reaction can be integrated into the process of antibody expression, which eliminates the need for using enzymes or protein-modifying agents during conjugation process.
To install fGly, a short consensus sequence — CXPXR, where X is usually serine, threonine, alanine, or glycine — is inserted at a desired location in the conserved regions of antibody heavy or light chains using standard molecular biology techniques. The resulting tagged construct is produced recombinantly in cells that coexpress FGE, which cotranslationally converts the cysteine within the tag into an fGly residue. That generates antibodies expressed with two aldehyde tags per molecule. The unique reactivity of the fGly is exploited to conjugate a cytotoxic payload through a Hydrazino-iso-Pictet-Spengler (HIPS) ligation that forms a stable carbon– carbon bond between payload and antibody (Figure 3c).
SMARTag technology has been used to create site-specific ADCs in which a maytansine payload was conjugated to different locations on an aldehyde tagged HER2 antibody. Serum stability experiments reveal that the HIPS chemistry seems to be less susceptible to protein microenvironment at different conjugation sites because the conjugation results in a carbon–carbon bond. However, the different conjugation sites influence the rate of conjugation, PK behavior, and efficacy in tumor xenograft models. Immunogenicity potential of antibodies with different tag placements and conjugation sites (as measured by an EpiScreen T-cell epitope mapping assay from Antitope, an Abzena company) was similar to that of untagged native antibody.
The Future of Site-Specific Conjugations
The R&D process for ADC drug development is an extremely complex, multiparametric optimization challenge. Early studies involving different site-specific methods clearly show that such conjugations can generate homogeneous ADCs. That will not only help with the process, characterization, and chemistry, manufacturing, and controls (CMC) aspects of production, but it can also improve the ADC therapeutic index and drive clinical differentiation.
Although most ADCs currently in clinical development rely on conventional conjugation chemistries, the first wave of site-specific ADCs is starting to enter clinical testing. If the early programs deliver clinical success, that will provide an even stronger push to embrace site-specific conjugations as the appropriate method for making optimized ADCs. Multiple technology options are available for site-specific conjugation, and adoption of a particular method will reflect a combination of commercial and technical considerations.
Studies with technologies that offer the option of flexible site-specific conjugations (THIOMAB, nnAA, TG, and aldehyde tagging) have firmly established that the site of conjugation significantly influences the pharmacological properties of an ADC. Thus it should be considered as a critical parameter in product design. Whereas it may seem that all this adds another criterion and complexity for ADC optimization, the ability to find an optimal site for a particular payload–antibody combination can be critical to the success of the program. As has been demonstrated, it can be easily implemented into preclinical research programs. It is likely to play a key role in the future of ADC development, making possible bispecific ADCs and helping companies develop ADCs for indications beyond oncology.
References
1 Biotech Products in Big Pharma Clinical Pipelines Have Grown Dramatically. Tufts CSDD Impact Report 15(6) 2013.
2 PhRMA 2013 Report. Overview, Medicines in Development: Biologics. Pharmaceutical Research and Manufacturers Assocation: Washington, DC, 2013; www. phrma.org/sites/default/files/pdf/ biologics2013.pdf.
3 Finco O, Rappuoli R. Designing Vaccines for the Twenty-First Century Society. Front. Immunol. 5(12) 2014: 1–6.
4 Veronese FM, Mero A. The Impact of PEGylation on Biological Therapies. Biodrugs 22(5) 2008: 315–329.
5 Sievers EL, Senter PD. Antibody– Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 64, 2013: 15–29.
6 Zolot RS, Basu S, Million RP. Antibody–Drug Conjugates. Nat. Rev. Drug Disc. 12(4) 2013: 259–260.
7 Wang L, et al. Structural Characterization of the Maytansinoid– Monoclonal Antibody Immunoconjugate, huN901–DM1, By Mass Spectrometry. Protein Sci. 14(9) 2005: 2436–2446.
8 Doronina SO, et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat. Biotechnol. 21(7) 2003: 778–784.
9 Sun MMC, et al. Reduction− Alkylation Strategies for the Modification of Specific Monoclonal Antibody Disulfides. Bioconjuate Chem. 16(5) 2005: 1282–1290.
10 Senter PD, Sievers EL. The Discovery and Development of Brentuximab Vedotin for Use in Relapsed Hodgkin Lymphoma and Systemic Anaplastic Large Cell Lymphoma. Nat. Biotechnol. 30(7) 2012: 631–637.
11 Hamblett KJ, et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 10(20) 2004: 7063–7070.
12 Junutula JR, et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 26(8) 2008: 928–932.
13 Junutula JR, et al. Engineered Thio- Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. Clin. Cancer Res. 16, 2010: 4769–4778.
14 Jeffrey SC, et al. A Potent Anti- CD70 Antibody–Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug with Site- Specific Conjugation Technology. Bioconj. Chem. 24(7) 2013: 1256–1263.
15 Shen B, et al. Conjugation Site Modulates the In Vivo Stability and Therapeutic Activity of Antibody–Drug Conjugates. Nat. Biotechnol. 30(2) 2012: 184–191.
16 Axup JY, et al. Synthesis of Site- Specific Antibody–Drug Conjugates Using Unnatural Amino Acids. Proc. Natl. Acad. Sci. 109(40) 2012: 16101–16106.
17 Zimmerman ES, et al. Production of Site-Specific Antibody–Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconj. Chem. 25(2) 2014: 351–361.
18 Strop P, et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 20(2) 2013: 161–167.
19 Rabuka D, et al. Site-Specific Chemical Protein Conjugation Using Genetically Encoded Aldehyde Tags. Nat. Protocols 7(6) 2012: 1052–1067.
20 Tian F, et al. A General Approach to Site- Specific Antibody Drug Conjugates. Proc. Natl. Acad. Sci. 111(5) 2014: 1766–1771.
21 Jackson D, et al. In Vitro and In Vivo Evaluation of Cysteine and Site Specific Conjugated Herceptin Antibody-Drug Conjugates. PLOS One 14 January 2014: 1–14.
22 Hallam TJ. Concept to Clinic in 365 Days? 3rd World ADC Summit, San Francisco, CA, 24 October 2012; http://adc-summit.com/uploads/ files/1972/Trevor_Hallam.pdf.
23 Strop P. Versatility of Microbial Transglutaminase. Bioconj. Chem. 25(5) 2014: 855– 862.
24 Jeger S, et al. Site-Specific and Stoichiometric Modification of Antibodies By Bacterial Transglutaminase. Angew. Chem. Int. Ed. Eng. 49(51) 2010: 9995–9997.
25 Dennler P, et al. Transglutaminase-Based Chemo-Enzymatic Conjugation Approach Yields Homogeneous Antibody–Drug Conjugates. Bioconj. Chem. 25(3) 2014: 569–578.
26 Dorywalska MG, et al. WO 2012059882: Engineered Polypeptide Conjugates and Methods for Making Thereof Using Transglutaminase. Pfizer Inc., Rinat Neuroscience Corporation; www.google. com/patents/WO2012059882A3?cl=en.
27 Drake PM, et al. The Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct In Vivo Efficacy and PK Outcomes. Bioconj. Chem. 25(7) 2014: 1331–1341. •
Corresponding author Abhijit S. Bhat is vice president of process development, and David Rabuka is founder and chief scientific officer of Redwood Bioscience, 5703 Hollis Street, Emeryville CA 94608; abhat@redwoodbioscience.com, drabuka@ redwoodbioscience.com, www.redwoodbioscience.com. Gregory Bleck is global head of biologics R&D at Catalent Pharma Solutions, 726 Heartland Trail, Madison WI 53717; gregory.bleck@catalent. com; www.catalent.com. Trademarks are the property of their respective owners.