Vaccines represent one of the most important medical developments in human history. As recently as a century ago, infectious diseases were the main cause of death worldwide, even in the most developed countries. For instance, the Spanish flu pandemic of 1918 killed more people than all the bullets and bombs did during World War I (1).
Today, a vast range of vaccines are available to protect against more than two dozen infectious diseases, especially in pediatrics. Our society has found that the only way to control or even eliminate certain diseases is consistent, widespread use of vaccines.
Conventional vaccines are generally, excellent generators of antibody responses. Vaccine-generated antibodies can bind to various outer parts of viruses, bacteria, or toxins circulating in the bloodstream and as such, prevent the pathogen from establishing an infection. However, this mode of action limits conventional vaccines to preventing infection only if an individual is immunized before initial exposure to a given pathogen.
Many of the most menacing infectious diseases in the world such as tuberculosis and malaria continue to elude conventional vaccine approaches. HIV and pandemic flu are still claiming millions of lives annually. Other challenging infectious diseases such as Chikungunya and dengu have the potential to wreak even greater havoc globally. It becomes clear that, like in other industries, we must change our approach to diseases new circumstances by using new technology.
Conventional vaccines use weakened or killed viruses or different parts of them as vaccines. Many are still grown in eggs or cells and harvested over weeks of time. To step beyond this practice, we are now pushing the frontiers with DNA vaccines, a new approach to vaccine design, formulation, manufacturing, and delivery.
DNA-Based Vaccines
DNA vaccines in clinical development today offer major benefits over their conventional counterparts (2,3). They are made up of specific strands of DNA that encode specific antigens and do not require isolated virus for production. Instead, DNA vaccines are delivered using expression plasmids, small circular strings of DNA that encode for a specific antigen or vaccine target. Unlike inactivated vaccines, they can mimic the immunological effects of infection. Because DNA vaccines directly transfect host cells, gene expression occurs through the host’s own machinery (e.g., transcription factors, ribosomes, and messenger ribonucleoprotein [mRNP]), thereby allowing for antigen presentation through both the major histocompatibility complex (MHC) class I and II pathways. By better mimicking the effects of natural viral infections, DNA vaccines can generate more powerful and broad immune responses than conventional vaccines. Unlike live-attenuated viral vaccines, however they pose zero threat for reversion to a virulent form. So they cannot replicate or cause disease. DNA remains the only vectored platform that does not induce antivector immunity, making it suitable for vaccine regimens that include both priming and boosts.
Manufacturing plasmid DNA is considerably faster and easier than most other vaccine platforms and relies primarily on bacterial hosts for production. Indeed manufacture of small-scale, non-GMP, research-grade plasmid material has become a commodity business. One advantage is that the difficulties associated with manufacturing and handling live/attenuated viral vaccines as well as large variability in potency from lot to lot are largely not an issue with DNA. Although the costs of current good manufacturing practice (CGMP) DNA manufacturing at small scale remain relatively high (on the order of $50–100/mg at a 1- to 10-g scale of manufacturing using 500-L fermentor process trains), the relative ease of plasmid DNA manufacturing and scale up makes it likely that future manufacturers will find ways to lower costs by 2–3 log10 at commercial scale (toward 10 kg using 3,000- to 30,000-L fermentor process trains) (4).
Although commercial scale-up with DNA has not been attempted — largely due to a lack of relevant late-stage vaccine products — the projected costs of manufacturing DNA at scale appear to be competitive with other licensed or other evaluated vaccine platforms such as live/attenuated virus, inactivated virus, VLP, or viral vectored (Ad5, MVA). Furthermore, unlike platforms that often require mammalian cell culture, DNA does not face the same risks with carryover of adventitious viruses (5,6), large variability in lot-to-lot potency, or safety of a live vialed final product. Because DNA vaccines are inherently stable, they also do not require cold-chain storage, which is a major logistical issue with some current conventional vaccines, especially in developing countries.
Perhaps the most important attribute is the potential of DNA vaccines to generate both robust antibody and T-cell responses. This ability means that DNA vaccination offers a therapeutic vaccination solution against many complex diseases such as HIV/AIDS and cancers.
And yet for all the promise, early DNA vaccine human clinical trials failed to meet immunogenicity end points. In test after test with early human trials, the one glaring shortcoming of this new vaccine technology was effective delivery of DNA plasmids into cells.
Electroporation Enhanced DNA Vaccine Delivery
In the 1970s, scientists discovered that applying electrical pulses to a cell in a petri dish enabled dramatically increased uptake of a biological material into the cell. Although electroporation (EP) was widely used as a laboratory technique, it wasn’t until the 1990s that the first research was undertaken to investigate potential direct applications of EP to humans in vivo. Over the past several years, Inovio Pharmaceuticals has pioneered development of in vivo EP for DNA vaccine delivery and commands a significant intellectual property position in this field.
Figure 1 shows how EP facilitates the uptake of useful molecules such as a DNA vaccine into a cell. After a DNA vaccine is injected into the target tissue, a brief, controlled electrical pulse is applied directly to that tissue. Just milliseconds of electroporation pulses temporarily open pores in the cell membrane, allowing a significant quantity of the previously injected DNA vaccine to enter cells. After a short period of time the pores reseal, leaving cells undamaged. By some measurements as much as 1,000-fold more DNA plasmids enter the cells by this method, leading to a 100- to 1,000-fold greater immune response (7,8,9).
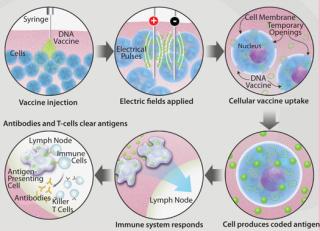
Studies have shown the potential of EP delivery of DNA vaccines in relevant nonhuman primate (NHP) models. Hirao et. al. evaluated the Merck Ad5 SIV vaccine — an important benchmark for new T-cell– based vaccine development — against an optimized SIV DNA vaccine delivered via the Cellectra EP device. They noted significantly higher immune responses, greater CD8+ T-cell responses, and increased polyfunctionality of CD4+ and CD8+ T-cells in the DNA vaccinated group compared with the Ad
5 group (10).
Another NHP study delivered an anthrax DNA vaccine with and without EP and compared efficacy in a challenge model to that achieved with a licensed anthrax vaccine (11). The authors reported a 100-fold enhanced immune response when the DNA vaccine was delivered via EP compared with standard intramuscular injection. The DNA EP vaccine conferred protection to the animals in a subsequent lethal Bacillus anthracis spore challenge comparable with that achieved with the licensed attenuated anthrax vaccine.
So although the lead DNA-EP vaccine programs are still only in phase 2, the weight of available data suggests that many of the desired goals for this platform are within reach and that the approach is likely to have a very bright future.
A DNA Vaccine Design
In vivo EP vaccine delivery may be the key enabling technology to finally make DNA vaccination clinically and commercially viable. The dramatic improvements achieved in DNA vaccine delivery and potency allows researchers to reach for the “Holy Grail” of vaccine development: designing vaccines that can protect against a broader range of evolving strains of diseases, including influenza and HIV, to which the vaccine is not specifically matched. This is an important leap beyond conventional vaccines, which must match the pathogen strain to provide protection.
Because DNA vaccines can combine genomics with in vivo antigen expression, they provide a tantalizing opportunity to easily customize vaccines by combining the strengths of molecular biology and genetic engineering to harness the potential of the immune system. The ability to combine multiple plasmids or disparate gene products into a single formulation without apparent loss of potency allows the possibility to formulate multicomponent vaccines targeting multiple antigens or even multiple pathogens simultaneously (12,13).
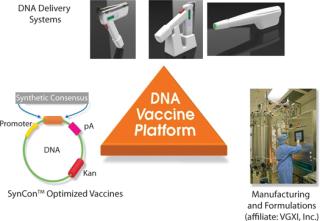
Inovio’s synthetic consensus vaccine design process begins with the alignment of a selected set of sequences for a given antigen and then identification of the most conserved amino acids at each position. The conserved amino acids are pieced together to yield a consensus immunogen. In theory, the consensus immunogen should contain the most conserved features of the component viruses while at the same time introducing some variability unique to the component sequences. By design, this unique process yields related but unmatched SynCon vaccines to “targeted” known and unknown virus strains. This is an important feature for targeting viruses that show high genetic diversity or high mutation frequencies because they pass from host to host. The SynCon vaccine design process is combined with potent EP delivery systems and efficient manufacturing processes, forming a vertically integrated DNA vaccine development platform (Figure 2).
Best-in-Class T-Cell Immune Responses in Clinical Studies
Data from two recently completed phase 1 clinical studies demonstrate the capabilities of this DNA vaccine development platform. In those trials SynCon DNA vaccines delivered with EP device generated best-in-class T-cell immune responses.
The first is a recently reported preliminary safety and immunogenicity study of VGX-3100, a HPV-16/18 E6 and E7 DNA–based candidate vaccine delivered via EP for cervical cancer and dysplasia therapy (14). No SAEs or vaccine-related Grade 3 or 4 AEs were reported. The antigen specific antibodies and T-cell ELISpots observed were higher than previous reports from prior studies of HPV poxviral, peptide or DNA vaccines (15,16), and these were observed even at low DNA doses. Subsequently, Inovio also reported that best-in-class levels of T-cell immune responses were shown in 14 of 18 total patients (78%). Those responses were durable up to over two years (at the latest time measured). Inovio is currently conducting a phase 2 clinical trial for VGX-3100, which is assessing the vaccine in women with CIN 2/3 or CIN 3 cervical dysplasias, the stages of abnormal cells preceding cervical cancer. The company expects to enroll 148 patients in 25 study centers in the United States, South Korea, South Africa, Australia, and Canada.
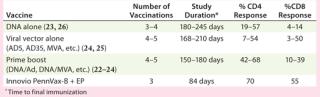
Similarly, strong T-cell immune responses were generated in a study (designated HVTN-080) involving vaccination of 48 healthy, HIV-negative volunteers to assess safety and levels of immune responses generated by Inovio’s Pennvax-B vaccine delivered with its Cellectra electroporation device in a randomized, double-blind, multicenter study conducted by the HIV Vaccines Trial Network (HVTN). Pennvax-B is a SynCon DNA vaccine that targets HIV gag, pol, and env proteins. All volunteers received the vaccine or placebo at months 0, 1, and 3. In an interim analysis, strong antigen-specific CD4+ T-cell responses were generated by the vaccine in 70% of evaluated vaccine recipients (21 out of 30). Similarly, strong antigen-specific, CD8+ T-cell responses were also generated by the vaccine in 55% of evaluated vaccine recipients (17 out of 31). Table 1 shows that the T-cell immune responses generated from that study were the best results from several dozen previous clinical studies conducted by the HVTN over the past two decades, including from those trials testing DNA vaccines without EP delivery, viral vector vaccines, and combinations using DNA and viral vectors.
Table 1: Comparison of T-cell immune responses from HIV vaccine trials
In terms of safety, DNA vaccines have been shown to have an excellent and consistently unremarkable safety profile. Safety trends have been revealed through more than 15 years of clinical development with plasmid-based DNA vaccines, collected across hundreds of human clinical trials, covering thousands of healthy and diseased subjects, spanning studies in infectious diseases, cancer, and gene therapy delivery. Such safety trends have continued to be observed in studies for which DNA has been delivered with EP. Published toxicology studies in animal models have largely yielded no adverse findings, and published human clinical studies have noted no vaccine-associated serious adverse events when DNA was administered as the drug substance either with or without EP (17,18,19,20). Tissue biodistribution studies in animals have noted that when found a
t all, DNA is present only at the injection site (usually skin and muscle). A rapid decay in plasmid copy numbers occurs over time (19,20), and early concerns surrounding plasmid integration into host genome remain unsubstantiated (21).
Results to date with DNA vaccines and EP have created a highly favorable safety profile. Although the safety database of DNA vaccines (with or without EP) is now at several thousand individuals (across hundreds of DNA vaccine trials) and growing rapidly, it still has not reached the maturity levels observed with the licensed vaccines. Thus potential concerns of long-term persistence and potential for integration will have to be addressed case by case.
The Decade of DNA Vaccines
This is an important “transitional” period for vaccines, for DNA vaccines delivered with electroporation technology, and for diseases we hope to prevent. The combination of highly optimized DNA delivered using advanced EP is clearly an important and exciting area of investigation. The amount of positive data from DNA–EP clinical studies support the notion that this is a vaccine product platform with broad applicability. At least half a dozen phase 2 clinical efficacy studies are on-going, including three trials Inovio is conducting alone and with its collaborators in the areas of cervical cancer, leukemia, and hepatitis C virus infection. Excitingly, the immune responses seen to date mimic or better those seen with viral infections in terms of the induction of both cellular and humoral responses and the magnitude and breadth of the responses. It is also encouraging that the scalability and the economics of manufacturing at scale allow the DNA vaccine platform to be competitive to other vaccine strategies for either therapeutic or prophylactic vaccine scenarios as well as their deployment in developed or resource poor settings. If these important developments continue to mount and additional successes are reported at the bench and in the clinic, we may well look back on the current time as the “decade of DNA vaccines.”
Author Details
J. Joseph Kim, PhD is cofounder and CEO at Inovio Pharmaceuuticals, Inc.; kim@inovio.com
REFERENCES