Cell therapies are being developed for a rapidly expanding range of indications. Genzyme Corporation has a treatment of cartilage in joints in long-term follow-up stage (Genzyme Corporation, www.genzyme.com). Cell Therapies are being investigated successfully in applications to treat infectious diseases such as AIDS, repair spinal cord injuries, strengthen immune systems, and treat neurological disorders such as Alzheimer’s disease, Parkinson’s disease, and epilepsy. Positive results have been reported in treating arteriosclerosis and other cardiovascular diseases, congenital defects, breast reconstruction, and liver insufficiency. Successes have also been reported in development of autologous cancer vaccines (1).
Developers of such products logically seek safety and efficacy in clinical trials. Strong positive results are highlighting the ever-widening fields of application and increasing numbers of registered clinical trials.
Contract Manufacturing Support of Clinical Trials: Many developers are using the services of contract manufacturing organizations (CMOs) to support clinical trials. Contract manufacturers offer proven facilities and process management that minimize the overhead to explore the competence of the therapeutic strategy. Additionally, CMOs have experience translating early R&D level processes into validated operations. This is particularly beneficial for small development companies because it facilitates clinical trial operations by outsourcing the actual manufacturing steps, managing regulatory requirements, conserving capital, and speeding up the development cycle.

It has often been said that “the product is the process” in biologics (2). Using a CMO in process development for a biologic enables that CMO to define the product in regulatory terms. In particular, long-term process implementation will be particularly defined through this early stage relationship because cell therapies are subject to the same regulatory oversight regarding manufacturing process changes as are other biotherapeutic products. In later development stages, establishing equivalence in manufacturing as it transits from manual to automated processing may require additional clinical studies to demonstrate a product’s continued safety, identity, purity, and potency.
Reliance on a CMO’s services has merit when you consider that CMOs succeed through intimate acquaintance with the ever-changing regulatory climate and through highly effective delivery, while minimizing investment costs. They are constantly alert for more efficient tools to make the job easier at all levels — from bench process to registration. It is reasonable to anticipate that a CMO experienced with the materials you are processing will systematically use best practices for the clinical trial level of activity.
Beyond Clinical Trials: If you look beyond, however, another picture emerges. During phase 3 trials of a cellular therapy, maybe 100 patients per year will be processed, perhaps two a week. If the process runs longer than two weeks (a common occurrence for cellular therapies), then it is probably acceptable to lock down cleanrooms for the duration of each patient lot. In such a case you would need four clean rooms to complete the trials without fear of cross contamination. Following registration, and on the assumption that there will be a commercially attractive patient population, you can expect annual process numbers to reach 1,000–10,000 patients. Is it realistic to continue the process in the same way? That would require a lot of cleanrooms! How about the personnel needed to work those rooms?

The Scale-Up Imperative: A critical issue with live cellular therapies is therefore the means to scale up production once registration is achieved. In-house process development and CMOs have no imperative to address this challenge. Access to the patient market and commercial returns from investment in the therapy are dramatically constrained without establishing a scalable manufacturing process within the clinical trial program.
What should such a manufacturing process look like? What are the key issues that need to be addressed? At Invetech, we have explored and developed processes directed at post-registration production for several cellular therapies and have identified some common conclusions.
QualityIf you have the chance to conduct or closely observe a manual cellular process, you will see many detailed manual operations: transferring fluids from vessel to vessel, taking cell counts, controlled dilutions, and so on. Under a validated process, every step is carefully monitored, as are any opportunities for operator-introduced variations.
Perspectives on Manual Processing: Bench-level cellular therapeutic production in the preclinical stage requires skilled and highly focused personnel working for long hours in cleanroom conditions. As many as three people may be involved: two operators and a quality supervisor verifying the batch record. Although this seems to be manageable when products for only one or two patients are in a facility at a time, is it practical to expect flawless processing when 40 patient products are being handled every week? From the perspective of a process failure risk, the probability of process failure is low when a focused regime can be sustained but the risk will increase as the frequency of processing increases.
The criticality of process failure risk can be very high. Batch failure of an autologous vaccine, for example, can cause a disastrous delay in treatment if a patient’s disease is progressing rapidly — representing a major risk. It may be considered unreasonable to expect process personnel to work under the pressure of that level of responsibility. Because of these quality and cost considerations, large-scale manual processing of these therapies becomes unviable at some point in scale.
Another aspect of manual processing is process consistency. People are wonderfully creative and take pride in doing their jobs well. The result is that different people complete the same task in subtly different ways. We all know that we have good and bad days. The fact is that operations performed manually will vary. Although the differences may seem trivial, if the process defines the product, then minor variations can result in substantially changed outcomes. Large-scale processing that contains steps subject to qualitative judgment should be regarded as unviable from a quality perspective.
Returning to the number of personnel required for a cellular therapy, the summary chart in Table 1 was created from a detailed study. These numbers are based on an eight-hour shifts per day, five-days per week operation modeled upon a real process. The skill requirements of the automated processing facility are substantially lower than the manual operations. It is evident that availability of skilled operators will affect the viability of manual processing from a centralized site.
Table 1: Personnel required for a cellular therapy

Table 1:
Successful registration of a therapy and growing sales will quickly challenge your logistics: obtaining patient materials on schedule, supporting clinicians, and delivering the materials to the administration site. Processing from a single centralized operation forces geographic constraints on sales. Transferring a validated manual process to external sites requires extensive training and careful revalidation against a new site’s operating protocols. It will probably be easier to replicate the organization of the originating site to minimize revalidation challenges created by variations in general protocols between sites. On this basis, each new location will effectively require investment in new facilities and creation of a management team and production personnel from a cold start.
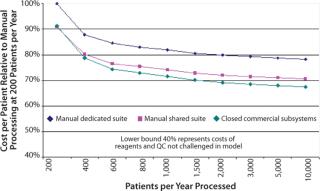
Cost of Therapy: The Commercial ImperativeAlthough the processing cost is relatively minor in the scheme of clinical trials, the commercial imperative requires a difference between the cost of delivering a service and reimbursement for providing it. Some efficiencies of scale can be expected with increasing patient numbers, but modeling the costs of manually processed therapies highlights the constraints on cost savings.
Many reagents and intermediate devices such as bead-based selection can add surprisingly to process costs. Good process design will ideally find multiple sources for reagents and consumables. This is not common at present, though in time some competitive purchasing may be possible. In the absence of this immediate opportunity, however, it is important to structure the process design to achieve the best cost directly and capture future cost reductions. The primary cost constraints that can be addressed are regulatory compliance, clean room capital, operating costs, and cost of labor.
Functionally Closed ProcessingMost cell therapy developers have recognized the benefits of operating in functionally closed, single-use vessels. Processes typically move in and out of closed systems through unit operations such as centrifugation, incubation, selection operations, washing operations, and so on. Devices for these activities are competently and rapidly developing, driven by repeat sales of the associated consumables.
Even though each unit operation is relatively automated, operators are required to present the materials to the equipment, verify the installation and program set up, and monitor the process. The good news is that a machine now replicates many of the “qualitatively controlled” manual processes such as vial-surface flushing during cell harvesting operations, which provides consistent and operator-independent quality.
The closed unit processes are commonly interlaced with “open” manipulations to collect samples, complete novel steps, formulation, and fill and finish, by way of example. These open steps necessitate operations in a sterile processing suite to address the risk of cross contamination. Sterile processing suites are expensive to maintain and cumbersome for personnel to work in. The cost of sterility verification for each batch is a major cost penalty. Gowning and cleaning also represent substantial labor and disposable costs.
To prevent cross contamination in a sterile processing suite, you have to eliminate all open processing steps. Sterile reagent preparation is not patient specific and can be conducted in a sterile suite shared with other nonpatient material preparation (subject to the usual risk evaluations of course).
With a process built around a complete set of functionally closed steps moving from instrument to instrument, it is possible to argue that operations in lower-class rooms will not place patients at greater risk. Indeed, operating in lower-class rooms means that personnel will be more comfortable and free to move in and out. You can also argue that integrity of the functionally closed systems also facilitates colocation of patient materials in the same room (Figure 1).
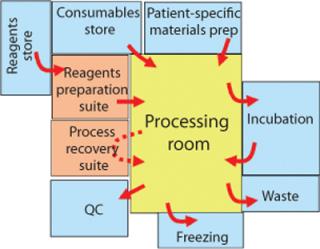
The benefits of achieving full segregation through functionally closed processing can be estimated by assuming shared lower grade rooms for processing and a small number of sterile processing suites for reagent preparation. As a mitigation plan for failures of a closed process, it is expedient to anticipate a sterile suite for manual recovery in proximity to a shared processing area.
A Sample Operation: With a functionally closed process in place, what will an operation look like? It is likely that two operators will still be required to complete set-up, processing, and clean-up of the process equipment. Close attention to detail will remain critical, as anyone who has worked a “Cytomate” in a formal process will verify. There are still many opportunities to get it wrong. A third person, as before, will be required to monitor the actions and verify the batch record. With multiple patient processes being conducted in one room, it may be possible for personnel to multitask to some degree, bounded by the risks this may introduce (Figure 2).
Managing Process Integrity: Even though unit operation devices using functionally closed disposables are steadily removing qualitative operator variance from these processes, there is still dependence on operator skill to reliably set up each operation and run it. The risks to be addressed in such activities include
-
connecting the correct materials in the right places at the right time before the process can proceed
-
monitoring every process step to verify that it is progressing correctly,
-
verifying that every process step has concluded successfully.
If you step through the activities of your process and apply this thinking to it, you will observe many gaps. For example, consider a stand-alone tube welder (Photo 1). How often have you completed a welding process to discover that the wrong ends of the tubes are in the wrong locations? If you then consider how you can be certain you have the correct tubes to connect at that time, then the process looks very ordinary. So, okay, we can read a barcode on each tube as it is presented to the welder. We can also place a physical interlock to ensure that the tube ends are placed correctly in the welder. But if the barcodes do not match, how do we prevent the welder from operating? Up till now, the only way to do this is place a physical restraint over the “weld” button. Commercial welders do not have an “inhibit” switch circuit that allows a control system to manage the operation.
Photo 1:
As a second example, consider the task of removing a sample midprocess for a cell count (Photo 2). To ensure that the correct sample is taken at the correct time in the process, the manually operated Sebra sealer is empowered by the process control system (www.sebra.com). At the appropriate time, an operator is prompted to remove the sample, and some visual cue such as flashing LEDs direct the operator to the sample to be removed. Orange dots in the illustration represent simple proximity sensors that advise the control system that the sealer has been presented to the cut point. If it is the correct cut point and time, the control system activates the sealer.
Photo 2:

The operator is unaware of this interlock unless he or she attempts the wrong action — in which case the sealer will not operate and the control identifies the incorrect action. Process integrity is sustained because no erroneous action took place. Once the sample has been removed, the control opens the pinch valve (illustrated as a green cylinder), thus opening the process line for the next operations. Systematically working through the entire process using a quality-by-design, risk-based approach delivers a process design with robust integrity.
Devices: Many stem-cell therapies are built around special devices for transport and administration of patient materials. Scaffolds and other implantable devices also figure within many cellular therapy concepts. Management of a process is required around these devices through to the administration/implementation actions to exploit these technologies.
Integrated AutomationIntegrated automation is a system comprising equipment platforms, disposables, and control systems that complete a specific process within functionally closed vessels, eliminating qualitative manual operations, and managing every aspect of process integrity. Integrated automation is motivated by the quality goals and commercial benefits it can deliver (Figure 3). The process design includes an equipment platform that supports disposable sets specifically designed for the application. These may include subsets of standard commercial disposables.

The benefits of a fully integrated system lie in the robust quality system it creates. The cost of therapy also benefits from a reduction in direct labor. Integration of disposable sets and full automated verification of set-up allows manual tasks to be conducted by a single operator with less dependence on long-term training. Design ownership of purpose-designed disposable sets enables management of disposable costs over the life of a therapy. The control system and disposable set configuration will be unique to each process.
Traceability and Batch Record Capture: With a fully integrated process control system and process monitoring to verify process integrity, the elements are in place to report process operations to a batch record. In its simplest form, this may be a printer that records the process steps and verification measurements. Heading the printout with a label linking to the patient ID links a process to its source and destination. Manually fitting labels to output packs sustains the path. By prelabeling the output packs with the state of the process they represent, the next process step is systematically identified.
This minimalist approach reflects an “islands of automation” strategy that can operate independently of a centralized manufacturing execution system (MES) or other communication structure. It enables relocation of the process with low infrastructure costs. The fully automated operation substantially minimizes training prerequisites and the impact of general standard operating procedures (SOPs) in a different facility.
Implementing an MES system may be beneficial as patient numbers grow and schedules must cover multiple geographic sites. Hooks for implementation of these organizational tools should be considered during design.
Cost of Therapy BenefitsFigure 4 illustrates the relative merits of integrated automation process initiatives. The semiautomated, closed system includes complete process integrity management in specifically design closed disposable sets. (The cost of development is included as an amortization over five years). Benefits arise from reduced operator labor costs. The fully automated closed system further benefits from automated batch record capture, hence eliminating process monitoring QA labor. The lower lines highlight the benefits of increased use of the capital by operating at 24 hours a day for seven days. The bottom line considers an aggressive production operation that is not burdened with development costs. This reflects the cost of therapy that a coprocessing partner would experience and provides some insight to the margin that can be reasonably applied by the process owner to cover the automated platform equipment and disposable development activity (Photo 3).


The integrated automation vision has stand-alone processing systems completing patient processes using purpose-designed consumable sets. Ownership of the platform, software, and consumable set design enables process owners to control their destinies and realize commercial opportunities that manually operated processes would struggle to sustain (Photo 4). Profound benefits include
-
product quality, process reliability, and integrity delivered through comprehensively monitoring every action
-
dramatic reduction in highly skilled staff needed to deliver commercial numbers of patient processes
-
stand-alone processing systems that enable rapid expansion of capacity to meet growing market
-
dramatic reduction in facility capital cost to encourage take-up by coprocessing partners
-
fully integrated process management and disposables supplied through the process owner to dramatically simplify establishment at new processing sites.

Table 2 and Figure 5 illustrate those capital cost reductions.
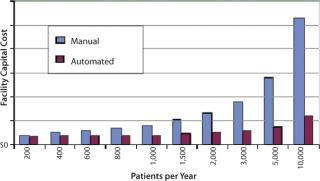
Table 2: Capital cost reductions (see

Ongoing Strengths: Ownership of single-use designs enables competitive sourcing into the future for significant cost reductions with increasing patient population as well as the essential guarantee of ongoing supply. Management of the disposable supply chain also facilitates centralized reagent sourcing, again creating the opportunity for future cost reductions. Process management and disposables supply through a centralized process owner facilitates ongoing control of quality and management of improvement initiatives across diverse sites.
A Hidden BarrierWe at Invetech have found that these observations are shared by many people and groups involved in cellular therapy development, particularly CMOs that anticipate the challenge they will confront when a therapy goes to market. CMOs also point out that many developing companies do not have the capital to progress this sort of thinking during clinical trials. Although it may be pragmatic in the short term, a point occurs in a clinical trial process at which the commercial viability of a therapy is locked in by formalization of the process. To quote the FDA, “changes in the manufacturing process, equipment or facilities may require additional clinical studies to demonstrate the product’s continued safety, identity, purity and potency” (2). Not addressing these issues directly will build a hidden barrier to commercialization, at the very least creating a systematic delay to commercial market realization.
When to ActIdeally, therapeutic process development is aligned with an automated processing strategy from the beginning. Failing this, once positive outcomes from phase 2 trials are emerging, it is important to position the process to enter phase 3 trials aligned for eventual automation. Introduction of fully automated processing within a phase 3 trial can then be navigated with the least potential disruption. Delaying beyond these times is likely to add development cost to emulate process steps not aligned for automation. In any event, phase 3 clinical trials need to be manufactured with a process that is representative of the final production approach. Constructing integrated automation solutions including disposables has a substantial labor component, and a period of two years should be anticipated for qualification of GMP systems. An early start to this activity with an advertised staged introduction during phase 3 has been accepted by regulators, in our experience (acknowledging that some extension to the phase 3 trial may be requested).
A Pragmatic Path: Use functionally closed processing strategies as soon as possible building on commercially available unit processing systems. In the commercialization plan for a cellular therapy, budget a process review for automation early in phase 2 trials. If a CMO is involved, its contribution to process design and compliance strategy will be important. Ongoing development work and early manufacturing operations are most likely to be conducted by the clinical trial processing organization. The outcome of each study should include a capital plan for scale-up, including development costs and tooling, to estimate a cost of therapy and drive commercial valuation of the initiative.
Breaking the Apron StringsEarly deployment of clinical trials with CMOs makes sense for many reasons. To a great extent, this relationship places much commercial initiative in the hands of the service company. To ensure that development is well positioned for commercial success, your strategy needs to anticipate a point at which separation from the service organization allows the full commercial potential of the your product to be realized. The integrated automation approach creates a vehicle for this transition into broad therapeutic application and commercial success.