To ensure product safety and efficacy, protein therapeutics must meet defined quality characteristics immediately after manufacture as well at the end of their designated shelf lives. Many physical and chemical factors can affect the quality and stability of biopharmaceutical products, particularly after long-term storage in a container–closure system likely to be subject to variations in temperature, light, and agitation with shipping and handling. Compared with traditional chemical pharmaceuticals, proteins are considerably larger molecular entities with inherent physiochemical complexities, from their primary amino acid sequences through higher-order secondary and tertiary structures — and in some cases, quaternary elements such as subunit associations (1).
Many proteins are glycosylated, and some have other posttranslational modifications such as phosphorylation, which also affects their potential degradation pathways as well as the kinetics of their degradation. Proteins are typically sensitive to slight changes in solution chemistry. They remain compositionally and conformationally stable only within a relatively narrow range of pH and osmolarity, and many require additionally supportive formulation components to remain in solution, particularly over time (2). Even lyophilized protein products experience degradation (3,4).
PRODUCT FOCUS: PEPTIDES AND PROTEINS
PROCESS FOCUS: FORMULATION/STABILITY
WHO SHOULD READ: ANALYTICAL AND FORMULATION DEVELOPMENT, PROCESS DEVELOPMENT, QA/QC AND REGULATORY AFFAIRS
KEYWORDS: FORCED DEGRADATION, PROTEIN DEGRADATION, OXIDATION, DISULFIDE SCRAMBLING, DEAMIDATION, AGGREGATION, HYDROLYSIS
LEVEL: INTERMEDIATE
Advances in analytical chemistry have identified many degradation pathways that can occur in recombinant protein therapeutics over time. These pathways generate either chemical or physical instability. Chemical instability refers to the formation or destruction of covalent bonds within a polypeptide or protein structures. Chemical modifications of protein include oxidation, deamidation, reduction, and hydrolysis (5).
Unfolding, dissociation, denaturation, aggregation, and precipitation are known as conformational or physical instabilities (5). In some cases, protein degradation pathways are synergistic: A chemical event may trigger a physical event, such as when oxidation is followed by aggregation.
Here, we present several protein degradation events: oxidation, photodegradation, disulfide scrambling, deamidation, aggregation, precipitation, dissociation, and fragmentation. We illustrate the biochemistry of each, showing potential means of induction and suggesting formulation considerations for prevention. In an upcoming issue, Part 2 will conclude with methods of detection and strategies for validation of stability-indicating methods. Our objective is to provide an introduction (or refresher) to the major degradation pathways of protein products, with references for each. Readers are encouraged to consult those references for expanded details on the basic biochemistry of each pathway, case studies describing experiments with specific proteins, and further information on formulation development strategies.
Oxidation, Photodegradation, and Disulfide Scrambling
Proteins and peptides are susceptible to oxidative damage through reaction of certain amino acids with oxygen radicals present in their environment. Methionine, cysteine, histidine, tryptophan, and tyrosine are most susceptible to oxidation: Met and Cys because of their sulfur atoms and His, Trp, and Tyr because of their aromatic rings (6). Oxidation can alter a protein’s physiochemical characteristics (e.g., folding and subunit association) and lead to aggregation or fragmentation. It can also induce potential negative effects on potency and immunogenicity depending on the position of oxidized amino acids in a protein relative to its functional or epitope-like domain(s).
For example, parathyroid hormone biological activity was differentially affected by a single oxidation of either Met-8 or Met-18 and double oxidation (Met-8 with Met-18) when each specie was isolated and testing using in vitro bioassays (7,8). Similarly, oxidation of Met-36 and Met-48 in human stem cell factor (huSCF) derived from Escherichia coli decreased its potency 40% and 60% (respectively) while increasing the dissociation rate constant of SCF dimer by two- to threefold, which suggests an effect on subunit binding and tertiary structure (9). In other cases, oxidation had no measurable impact on protein potency even when substantial structural changes were seen. For example, oxidized Met-111 in interferon α-2b affected the molecule’s primary, secondary, and tertiary structure and prevented site-specific epitope recognition by a monoclonal antibody (MAb) without altering in vitro biological activity (10).
Mechanism and Factors Involved: Figure 1 shows biochemical pathways for oxidation of methionine and cysteine residues. Methionine is oxidized by atmospheric oxygen and oxygen radicals in solution to form methionine sulfoxide and methionine sulfone. Both species are larger and more polar than nonoxidized methionine, which can alter protein folding and structural stability (11). The rate of methionine oxidation in recombinant human parathyroid hormone (rHu-PTH) by hydrogen peroxide is enhanced at alkaline pH (8).
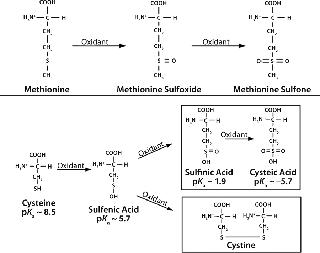
Cysteine oxidation is also more prevalent at alkaline pH, which deprotonates thiol groups. Oxidation of cysteine induces disulfide bond breakage in a reducing environment (Figure 1, BOTTOM). In such an environment, cysteine oxidation involves nucleophilic attack of thiolate ions on disulfide bonds, generating new disulfide bonds and different thiolate ions. The new thiolate can then react with another disulfide bond to form cysteine.
Such intermolecular disulfide links formed by protein degradation accumulate mispaired disulfide bonds and scrambled disulfide bridges, which can alter protein conformation and subunit associations (6). Cysteine residues may also undergo s
pontaneous oxidation to form molecular byproducts — sulfinic acid and cysteic acid — in the presence of metal ions or nearby thiol groups (11). For example, human fibroblast growth factor (FGF-1) exhibits copper-catalyzed oxidation that can create homodimers (12).
Spatial orientation of thiol groups in proteins plays an important role in cysteine oxidation. The rate of oxidation is inversely proportional to the distance between those thiol groups (13). This can eventually lead to formation of large oligomers or nonfunctional monomers, as with basic fibroblast growth factor (bFGF), which contains three cysteines that are easily oxidized and form intermolecular or intramolecular disulfide bonds (13).
This oxidation often induces conformational modifications of the protein because cysteine disulfide increases side-chain volume in the protein’s interior and leads to unfavorable van der Waals interactions that maintain the original structure (13).
Histidine residues are highly sensitive to oxidation through reaction with their imidazole rings, which can subsequently generate additional hydroxyl species (6). Oxidized histidine can yield asparagine/aspartate and 2-oxo-histidine (2-O-His) as degradation products during light and/or metal oxidation (6,14). It may be a transient moiety because it can trigger protein aggregation and precipitation, which can obscure isolation of 2-O-His as an individual degradant (15). Oxidation of tyrosine may result in covalent aggregation through formation of bityrosine (16). Spatial factors may also affect tyrosine and histidine oxidation. Adjacent negatively charged amino acids accelerate tyrosine oxidation because they have high affinity to metal ions, whereas positively charged amino-acid residues disfavor the reaction (17,18). If an adjacent amino acid is bulky, it may mask oxidation of neighboring amino acids and prevent them from getting oxidized. It has been observed that histidine present in a sequence markedly increases both the peptide oxidation rate and methionine sulfoxide production. The strong metal binding affinity of the imidazole ring on the histidine side chain brings oxidizing species close to the substrate methionine (6).
Light Degradation: Photooxidation can change the primary, secondary, and tertiary structures of proteins and lead to differences in long-term stability, bioactivity, or immunogenicity (19). Exposure to light can trigger a chain of biochemical events that continue to affect a protein even after the light source is turned off. These effects depend on the amount of energy imparted to a protein and the presence of environmental oxygen. Photooxidation is initiated when a compound absorbs a certain wavelength of light, which provides energy to raise the molecule to an excited state. The excited molecule can then transfer that energy to molecular oxygen, converting it to reactive singlet oxygen atoms. This is how tryptophan, histidine, and tyrosine can be modified under light in the presence of O2 (6). Tyrosine photooxidation can produce mono-, di-, tri-, and tetrahydroxyl tyrosine as byproducts (18). Aggregation is observed in some proteins due to cross-linking between oxidized tyrosine residues (20). Photooxidation reaction is predominately site specific (21). For example, in human growth hormone treated with intense light, oxidation is carried out predominantly at histidine-21 (22). In addition, the peptide backbone is also a photodegradation target (23). Alternatively, the energized protein itself can react directly with another protein molecule in a photosensitized manner, typically via methionine and tryptophan residues at low pH (6).
Excipients and leachables can synergistically affect the oxidation (and hence, degradation) of a protein.
Formulation components influence the rate of photooxidation in some instances: e.g., phosphate buffer accelerates the rate of methionine degradation more than other buffer systems do (22). Metal-ion–catalyzed oxidation depends on concentration of metal ions in the environment. The presence of 0.15-ppm chloride salts of Fe3+, Ca2+, Cu2+, Mg2+, or Zn2+ does not affect the rate of oxidation for human insulin-like growth factor-1, but when the metal concentration increased to 1 ppm, a significant increase in oxidation was observed (23). Oxidation can be exacerbated in the presence of a reducing agent such as ascorbate. Ascorbic acid increased oxidation of human ciliary neurotrophic factor (24). Also, the presence of denaturing/unfolding reagents in solution can increase the extent of protein oxidation. Excipients such as polyols and sugars involved in stabilizing protein structure can decrease the rate of oxidation (6).
Oxidative modification depends on intrinsic structural features such as buried and exposed amino acids. In the case of human growth hormone, Met-14 and Met-125 are readily oxidized by H2O2 because they are exposed to the surface of the protein, whereas Met-170 in its buried position can be oxidized only when the molecule is unfolded (21). Also, atmospheric oxygen can cause protein oxidation over time. Headspace oxygen contributed to the loss of 50% potency by four months in multidose vials of tuberculin purified protein (TPP) (25).
Oxidation can be induced during protein processing and storage by peroxide contamination resulting from polysorbates and polyethylene glycols (PEGs), both commonly used as pharmaceutical excipients. A correlation has been observed between the peroxide content in Tween-80 and the degree of oxidation in rhG-CSF, and peroxide-induced oxidation appeared more severe than that from atmospheric oxygen (26). Peroxide can also leach from plastic or elastomeric materials used in primary packaging container–closure systems, including prefilled syringes (27,28).
Preventive Measures: One molecular engineering strategy for minimizing oxidative degradation is to replace oxygen-labile amino acids with oxygen-resistant ones if a protein’s nature permits. In therapeutic Interferon beta (IFN-β), cysteine at position 17 was replaced by serine, because the former loses antiviral activity during storage to oxidation and disulfide scrambling (29). Substitution of methionine of epidermal growth factor (EGF) with a nonnaturally occurring norleucine also prevented oxidative degradation (30).
Removal of headspace oxygen by degassing may be effective for preventing oxidation in some cases. Filling steps are carried out under nitrogen pressure, and vial headspace oxygen is replaced with an inert gas such as nitrogen to prevent oxidation (21,25). With some oxidation-sensitive proteins, processing is carried out in the presence of an inert gas such as nitrogen or argon. For multidose drug preparations, use of cartridges with negligible headspace overcomes oxidation and related consequences (25).
Care must be exercised when container–closure changes are considered. Many such changes for protein therapeutics (from vials to prefilled syringes or prefilled syringes to pen devices, for example) are considered to enhance patient convenience and ease of use. But historical experience with container–closure systems based only on chemical ph
armaceuticals should be reevaluated when the same materials are used with protein-based products because of potential for unexpected, unique impacts on protein degradation.
Controlling or enhancing factors such as pH, temperature, light exposure, and buffer composition can also mitigate the effects of oxidation by affecting a protein’s environment. Cysteine oxidation often can be controlled by maintaining the correct redox potential of a protein formulation, such as with addition of thioredoxin and glutathione. Antioxidants and metal chelating agents also can be used to prevent oxidation in protein formulations. Antioxidants are chemical “sacrif icial targets” with a strong tendency to oxidize, consuming chemical species that promote oxidation. Scavengers such as L-methionine and ascorbic acid are used for this purpose in biotherapeutic formulations (31). In the absence of metal ions, cysteine as a free amino acid may act as an effective antioxidant. As chelating agents, EDTA and citrate might form complexes with transition metal ions and inhibit metal-catalyzed, site-specific oxidation (6). Addition of sugars and polyols may also prevent metal-catalyzed oxidation because of their complexion with the metal ions. Protective effects of glucose, mannitol, glycerol, and ethylene glycol against metal-catalyzed oxidation has been observed with human relaxin (32). Physical protection from UV/white light exposure with either a primary or secondary packaging system may be necessary to protect light-labile proteins from photooxidation.
Deamidation
With many recombinant proteins, changes in peptide and protein structure are observed through the nonenzymatic deamidation of glutamine and asparagine residues. This can have varying effects on their physiochemical and functional stability (33,34). It has been observed that deamidation of hGH alters proteolytic cleavage of the human growth hormone (33). And it was reported that deamidation of IFN-beta increased its biological activity (35). It has been determined that deamidation of peptide growth-hormone–releasing factor leading to aspartyl and iso-aspartyl forms reduces the bioactivity by 25- and 500-fold, respectively, as compared with the native peptide (36). Deamidation at an Asn-Gly site in hemoglobin changes its affinity to oxygen (37). Asparagine deamidation perturbs antigen presentation on Class II major histocompatibility complex molecules (38). It was reported that isomerization of Asp 11 in human epidermal growth factor led to a fivefold reduction in its mitogenic activity (39). And deamidation at two Asn-Gly sequences in triose-phosphate isomerase resulted in subunit dissociation (40).
Mechanism and Factors Involved: Deamidation is a chemical reaction in which an amide functional group is removed from an amino acid. Consequences include isomerization, racemization, and truncation of proteins. Figure 2 shows the mechanism of asparagine degradation by deamidation.
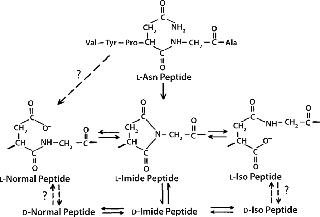
Isomerization: Isomerization of aspartate to isoaspartate residues in a protein solution is the most commonly observed outcome of nonenzymatic deamidation (41,42).
Racemization: Succinimide intermediates formed during asparagine deamidation are highly prone to racemization and convert to D-asparagine residues (41,42). Racemization of other amino acids, except glycine, is observed at alkaline pH (42).
Truncation: At low pH levels, peptides and proteins experience deprotonation of the amide group on their asparagine side chain, followed by nucleophilic attack by the nitrogen atom of the amide anion on the peptide carbonyl carbon of the asparagine residue (43). This generates peptide chain cleavage by forming a succinimide peptide fragment. Subsequent hydrolysis of the succinimide ring can yield asparaginyl and β-asparaginyl peptides.
Mechanisms for the aspartate–isoaspartate deamidation and isomerization reactions are similar because they both proceed through an intramolecular cyclic imide intermediate (44). Deamidation rates for individual amide residues depends on their primary sequence and three-dimensional (3D) structure as well as solution properties such as pH, temperature, ionic strength, and buffer ions (45). The deamidation rate for glutamine residues is usually less than that of asparagine residues (46,47).
If pH is >5.0, deamidation occurs through very unstable cyclic imide intermediate formation, which spontaneously undergoes hydrolysis. Under strong acidic conditions (pH 1–2), direct hydrolysis of the amide side chain becomes more favorable than formation of cyclic imide (48). Peptide bond cleavage occurs to a greater extent in direct amide hydrolysis. At neutral pH, deamidation can lead to structural isomerization.
The rate of deamidation is also influenced by protein secondary structure. Increasing helical structure decreases the rate of deamidation in some proteins (50). The rate of deamidation in several growth-hormone–releasing factor analogs was examined as a function of methanol-induced α-helical structure. Addition of methanol increased the level of α-helicity and decreased the rate of deamidation (51). In its native structure, RNAase resists deamidation possibly because of the relatively rigid backbone in the loop stabilized by a disulfide between Cys-8 and Cys-12 and by the β-turn at residues 66–68, which could hinder the formation of the cyclic imide (52). But if it is reduced and denatured, then refolded, aspartic and isoaspartic forms are generated, demonstrating different enzymatic activities. Replacement of Asp-67 with Iso-Asp-67 showed that the isoaspartic form refolds at half the rate of the fully amidated form (51).
Storage temperatures can affect a protein’s deamidation rate in the presence of certain biological buffers. Because amine buffers (e.g., Tris and histidine) have high temperature coefficients, storage at temperatures that are different from the temperature of preparati
on could shift formulation pH. Deamidation and isomerization reactions are pH-sensitive processes, so those shifts in formulation pH could alter the rate of deamidation. Another indirect effect of temperature is the dissociation constant of water: The hydroxyl ion concentration of water can vary as a function of temperature and thereby affect deamidation rates (39).
Preventive Measures: Solution pH can substantially affect deamidation (44). Formulations at pH 3–5 can minimize peptide deamidation (48). AsnA-21 and AsnB-3 of insulin forms isoaspartate or aspartate, depending on the pH of solution (52). Insulin deamidates rapidly at Asn A-21 in low pH solutions (57). Steric hindrance also can affect deamidation rate: Bulky residues following asparagine may inhibit the formation of succinimide intermediate in the deamidation reaction (42). Replacement of a glycine residue with more bulky leucine or proline residues resulted in a 30- to 50-fold decrease in the rate (42). In lyophilized formulations, the deamidation rate is typically reduced, probably due to limited availability of free water in which the reaction can occur.
Formulations that incorporate organic cosolvents can decrease their deamidation rates because addition of organic solvents decreases dielectric constants of a solution (44). Decreasing solvent dielectric strength — by addition of cosolutes such as glycerol, sucrose, and ethanol to a protein solution — leads to significantly lower rates of isomerization and deamidation (39,44). Lowering dielectric strength of the medium from 80 (water) to 35 (PVP/glycerol/water formulations) led to about a sixfold decrease in peptide deamidation rates (54). The lower rate of deamidation was attributed to less stabilized ionic intermediates formed during cyclization in the asparagine deamidation pathway. Insulin prepared in neutral solutions containing phenol showed reduced deamidation probably because of its stabilizing effect on the tertiary structure (α-helix formation) around the deamidating residue, which lowered the probability for formation of intermediate imides (53).
Aggregation and Precipitation
Aggregated proteins are a significant concern for biopharmaceutical products because they may be associated with decreased bioactivity and increased immunogenicity. Macromolecular protein complexes can trigger a patient’s immune system to recognize the protein as “nonself ” and mount an antigenic response (55). Large macromolecular aggregates also can affect fluid dynamics in organ systems such as eyes (56).
Aggregation is a common problem encountered during manufacture and storage of proteins (16). The potential for aggregated forms is often enhanced by exposure of a protein to liquid–air, liquid–solid, and even liquid–liquid interfaces (57). Mechanical stresses of agitation (shaking, stirring, pipetting or pumping through tubes) can cause protein aggregation. Freezing and thawing can promote it as well. Solution conditions such as temperature, protein concentration, pH, and ionic strength can affect the rate and amount of aggregates observed. Formulation in sucrose can increase aggregation over time because of protein glycation when sucrose is hydrolyzed (58). The presence of certain ligands — including certainions — may enhance aggregation. Interactions with metal surfaces can lead to epitaxic denaturation, which triggers aggregate formation. Foreign particles from the environment, manufacturing process, or container–closure system (e.g., silicone oil) can also induce aggregation (21,59,60). Even handling protein products at compounding pharmacies can induce aggregation 10-fold above initially observed amounts (58).
The impact of aggregation on product potency varies based on the physiochemical attributes of each protein relative to its functional domains and the nature of the activity being measured. Enzymes such as urease and catalase can lose up to 50% of their potency after shaking, fibrinogen clotting activity is decreased after shear stress, and recombinant IL-2 and recombinant interferon activity is substantially affected by aggregation from shaking and shearing (2). Aggregation also affects the mass balance of protein solutions, decreasing the concentration of the target protein. Microaggregated subvisible particles generated anywhere in a manufacturing process can develop into larger particles over time as a product is stored (62). Bevacizumab drug product lost 50% of active IgG after manipulations at the pharmacy triggered significant growth of micron-sized particles in repackaged solutions (58).
Aggregates can be soluble or insoluble, reversible or irreversible, covalent or noncovalent (16). Soluble aggregates are usually reversible: e.g., by altered solution conditions such as changing temperature or osmotic strength or by mild physical disruption such as swirling or filtration. Insoluble aggregates are typically irreversible. Under vigorous physical disruption (e.g., agitation or freezing and thawing) or over time in storage, they can grow into particles that may eventually precipitate. Covalent aggregates form when monomeric proteins become chemically crosslinked, e.g., though disulfide bonds. Although covalent linkages are necessary to stabilize the native tertiary structure of most polypeptide proteins, those that form by degradation can produce undesired crosslinks between protein moieties, which can lead to irreversible aggregation. Noncovalent aggregates are formed when proteins associate and bind based on structural regions of charge or polarity. Because such associates are weak (relative to covalent linkages), they are sensitive to solution conditions and usually reversible.
Mechanism and Factors Involved: Because of the many physical and chemical manipulations required in upstream production and downstream processing, followed by formulation and filling operations, aggregation of protein biopharmaceuticals can be induced during nearly every step of the process including at hold points, shipping, and long-term storage (16,21). Agitation (e.g., shaking, stirring, and shearing) of protein solutions can promote aggregation at the air–liquid interfaces, where protein molecules may align and unfold, exposing their hydrophobic regions for charge-based association (2). Agitation-induced aggregation has been seen in numerous protein products, including recombinant factor XIII, human growth hormone, hemoglobin, and insulin (2). Minimizing foaming caused by agitation during manufacture (as well as during product use) may be critical to preventing significant loss of protein activity or generation of visible particulate matter (62).
Protein concentration also can promote aggregation, with or without agitation events. Results obtained from two PEGylated proteins and one Fc fusion protein demonstrated a direct correlation between protein concentration and aggregation under nonagitated (quiescent) conditions, but researchers found an inverse correlation between protein concentration and aggregation under conditions of shaking, vortexing, and simulated shipping (63).
Antimicrobial preservatives used in multidose formulations also can induce protein aggregation. For example, benzyl alcohol accelerates the aggregation of rhGCSF because it favors partially unfolded conformation
s of the protein (64). Increasing antimicrobial preservative levels may increase the hydrophobicity of a formulation and could affect a protein’s aqueous solubility (62).
Phenol and m-cresol can considerably destabilize a protein: Phenol promotes formation of both soluble and insoluble aggregates, whereas m-cresol can precipitate protein (65).
Freezing and thawing — which can occur multiple times throughout production and use of protein therapeutics — can dramatically affect protein aggregation. Generation of water-ice crystals at a container’s periphery (where heat transfer is greatest) can produce a “salting out” effect, whereby the protein and excipients become increasingly concentrated at the slower-freezing center of a container (21). High-salt and/or high-protein concentrations can result in precipitation and aggregation during freezing, which is not completely reversible upon thawing. The effect can be seen with thyroid-stimulating hormone: When stored at −80 °C, 4 °C, or 24 °C for up to 90 days, it remained stable, but when frozen to −20 °C it lost >40% potency in that period, which was attributed to subunit dissociation (66). Multiple freezing and thawing cycles can exacerbate that effect and lead to a cumulative impact on the generation and growth of subvisible and visible particulates. A change in pH can come from crystallization of buffer components during freezing. In one study, potassium phosphate buffers demonstrated a much smaller pH change on freezing than did sodium phosphate buffers (21).
Compendia currently limit the number of particles ≥10 μm and ≥25 μm in size that may be present in injectible pharmaceutical preparations (67,68). However, what levels of subvisible particles (2). Also, no standards are codified for visible particulates in protein pharmaceutics. Some biotechnology products have visual appearance specifications for drug solutions that include comments such as “essentially free of visible particulates” or “some translucent particles may be present” (69).
Preventive Measures: A protein solution typically can be stabilized against aggregation and precipitation by optimizing solution pH and ionic strength; adding sugars, amino acids, and/or polyols; and using surfactants (70). Comprehensive evaluation of optimal pH and osmotic conditions is a key element of formulation development to prevent protein aggregation or precipitation (71). Irreversible aggregation due to denaturation can be prevented with surfactants, polyols, or sugars (62).
In many cases, nonionic detergents (surfactants) are added to increase stability and to prevent aggregation. The protein–surfactant interaction is hydrophobic, so these compounds stabilize proteins by lowering the surface tension of their solution and binding to hydrophobic sites on their surfaces, thus reducing the possibility of protein–protein interactions that could lead to aggregate formation (72). The nonionic detergents Tween 20 and Tween 80 can prevent formation of soluble protein aggregates with surfactant concentrations below the critical micelle concentrations (CMC) (73). Polysorbate (Tween) 80 added to IgG solutions stabilized small aggregates and prevented them from growing into larger particles (74). Chelating agents also can be used to prevent metal-induced protein aggregation (62,70).
Fragmentation
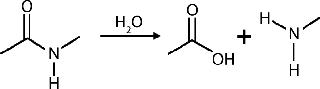
Multimeric proteins with two or more subunits can become dissociated into monomers, and monomers (or single peptide chain proteins) can degrade into peptide fragments. Nonenzymatic fragmentation usually proceeds by hydrolysis of peptide bonds between amino acids, releasing polypeptides of lower molecular weight than the intact parent protein (75). Peptide bonds of Asp–Gly and Asp–Pro are most susceptible to hydrolytic protein cleavage (75,76). Antibody hydrolysis often occurs at the hinge region, which is the most flexible domain of an antibody (77).
However, decreasing pH from 9 to 5 can shift the peptide hydrolysis sites of a recombinant monoclonal antibody, showing increased cleavage outside that region (78).
The presence and position of oligosaccharides also affects the rate of peptide hydrolysis at low pH levels. Depending on location, hinge-region cleavage was not affected although fragmentation in the CH2 domain decreased (77). Hydrolytic cleavage of peptide bonds by acidic and basic hydrolysis are not necessarily the same effects. Recombinant human macrophage colony-stimulating factor yielded different peptide fragments in solutions with acidic and basic pH (74). Enzymatic protein fragmentation can be caused by proteolytic activity of residual or contaminating proteases — or in select cases, autoproteolysis of an enzymatic protein (79).
Preventive Measures: Appropriately buffering formulations to maintain their solution pH in a suitable range for each protein type is key to minimizing hydrolytic fragmentation. For example, calcitonin undergoes hydrolysis at basic pH, but no such degradation is observed at pH 7 even at room temperature (80). Buffer composition may also affect hydrolysis. Recombinant human macrophage colony-stimulating factor fragmentation was observed in phosphate-buffered solutions but not in histidine-buffered solutions at identical pH and ionic strengths (76). It is also important to minimize the potential presence of proteases in protein purification, either from sources intrinsic to the production process (e.g., host-cell proteins) or from extrinsic sources of contamination (e.g., adventitious microbes).
The native structure of a protein molecule is the result of balancing effects such as covalent linkages, hydrophobic interactions, electrostatic interactions, hydrogen bonding, and van der Waals forces (1). Protein stability is controlled by innumerable intrinsic and extrinsic factors, but the major ones are primary sequence, 3D structure, subunit associations, and posttranslational modifications (2). Extrinsic contributing factors include pH, osmolarity, protein concentration, formulation excipients, and exposure of a product to physical stress from temperature, light, and/or agitation. Leachables from container–closure systems and contamination from the environment (e.g., metals and proteases) also exacerbate product degradation. Taken together, all these features make protein degradation a very complex physiochemical phenomenon, so formulation optimization is a key aspect of biotechnology product development.
Author Details
Jalpa Patel and Ruchi Kothari are research associates, Rashbehari Tunga is a principal scientist, and corresponding author Binita S. Tunga is senior scientist in the stability study laboratory in R&D at Intas Biopharmaceuticals Ltd., Plot No. 423/P/A/GIDC, Serkhej Bavla Highway, Moraiya-382210, Ahmedabad, India; 91-2717-660100; fax 91-2717-251189; tunga72@hotmail.com; www.intasbiopharma.co.in. Nadine M. Ritter is senior consultant of Biologics Consulting Group (Alexandria, VA) and a member of BPI’s editorial advisory board.
REFERENCES
ys. 248:452-459.
Could you please send me the link for the Part 2 of the above article? We tried hard but couldn’t find it the Archive as well as through the search function of this website. Your help would be highly appreciated. Many thanks in advance.
Unfortunately, the second part of this article was never published. If you have specific stability questions, you may want to reach out to Nadine Ritter, President and Analytical Advisor at Global Biotech Experts. She helped with the publication of this article and has significant experience in this area.
Also, I have removed reference to Part 2, so that others may avoid the confusion in searching for it. Thanks!