 The reprogramming of human somatic cells into induced pluripotent stem cells (iPSCs) offers tremendous potential for cell therapy, basic research, disease modeling, and drug development. Human iPSCs can be generated in culture, expanded, and then used to manufacture clinical-grade cells of almost any adult cell type. Given their great capacity for self-renewal, they are attractive as input materials for current good manufacturing practice (CGMP) operations.
The reprogramming of human somatic cells into induced pluripotent stem cells (iPSCs) offers tremendous potential for cell therapy, basic research, disease modeling, and drug development. Human iPSCs can be generated in culture, expanded, and then used to manufacture clinical-grade cells of almost any adult cell type. Given their great capacity for self-renewal, they are attractive as input materials for current good manufacturing practice (CGMP) operations.
For human iPSCs to fulfill their therapeutic potential, however, it is necessary to develop a CGMP- compliant iPSC manufacturing process and address challenges related to tissue sourcing, characterization, and testing of iPSC banks. Here, we summarize an approach to address the logistic and technical challenges involved in manufacturing clinical-grade cells. In addition to establishing a tissue- sourcing protocol, we developed a defined, feeder-free, and xeno-free reprogramming and cell culture system that supports efficient generation and expansion of human iPSCs. By incorporating proper process design considerations, we optimized the process and then transitioned it into a CGMP facility where human iPSCs were generated in CGMP compliance and tested with a well-defined quality control (QC) and testing strategy.
Both cell lines we manufactured fulfilled the safety, pluripotency, and identity criteria for high-quality, clinical-grade human pluripotent stem cells. These cell lines can be used later to generate specialized cells for different clinical applications.
Background
Isolation of human embryonic stem cells (hESCs) from the inner cell mass of eight-day-old blastocysts provided a pluripotent starting material (able to form all cell types derived from ectoderm, endoderm, and mesodermal lineages) (1). This remarkable accomplishment has dramatically changed the fields of developmental biology, in vitro differentiation, and regenerative medicine.
In 2007, Shinya Yamanaka successfully converted adult human cells to iPSCs (2). That gave them similar characteristics to embryonic stem cells (ESCs). By definition, iPSCs can indefinitely self-renew and become any other anatomical cell type. The process of reprogramming with retroviruses that express four transcription factors (Oct3/4, Sox2, Klf4, and c-Myc) was readily replicated worldwide and improved upon by other investigators. Similar to ESCs, human iPSCs are pluripotent and can be readily derived from any donor. They have become an important scientific tool spurring advancements in basic research, disease modeling, drug development, and regenerative medicine. Equally important, this discovery unlocked many new opportunities for both allogeneic and autologous cell therapy applications.
The first iPSC lines were reprogrammed using retrovirus constructs, which permanently integrated into the cells’ genome and thus are not preferred for generating clinically relevant cell populations for therapeutic applications. Moreover, cells were usually generated and expanded using a feeder-layer system, which incurs lot-to-lot variability, regulatory and safety concerns, and scalability problems.
An alternative to that cumbersome and risky process is to use nonintegrating plasmid DNA carrying the same or similar transcription factors into somatic cells, such as CD34+ cells derived from newborn umbilical-cord blood or adult peripheral blood mononuclear cells (3, 4). However, switching to integration- free methods and potentially clinically compliant methods to generate human iPSCs has been relatively inefficient and technically challenging.
Those realities led Lonza to focus on developing a robust, reproducible, and CGMP-compliant manufacturing process to generate clinical grade iPSCs for use in manufacturing therapeutic cellular products. Here, we summarizes our approach to developing such a process. We hope it can serve as a guide for readers interested in manufacturing iPSCs for therapeutic use.
Process Requirements and the Path to CGMP
We took a step-by-step approach to establish a robust, reproducible, CGMP-compliant iPSC manufacturing process. As Table 1 shows, the steps first established an iPSC generation process using a nonintegrating, episomal-based technology in Stage 1 (proof of principle). In Stage 2, we performed process optimization and protocol development based on critical process attributes. And in Stage 3, we transferred that manufacturing process into a CGMP cell therapy suite. We considered a number of design inputs in development of this iPSC manufacturing process:
- iPSC derivation (e.g., safety and efficiency of the reprogramming method, donor-to-donor variability, and choice of starting materials)
- iPSC challenges (e.g., development of a cell culture system for iPSC generation and expansion, cell sensitivity and robustness, and cryopreservation and revival)
- safety and QC challenges (e.g., normal karyotype, residual plasmid clearance, in-process controls to evaluate iPSC quality, critical attributes of the final products, and standard safety concerns such as sterility).
Other challenges include labeling and packaging, final-product storage and warehousing, facilities, human resources, and training. We also considered equipment and utilities during CGMP manufacturing process design. In addition to incorporating design considerations related to iPSC manufacturing; we carefully considered and evaluated regulatory issues applicable to tissue acquisition and iPSC manufacturing and testing from the early process stages. Detailed discussion of these challenges and their solutions is beyond the scope of this discussion.
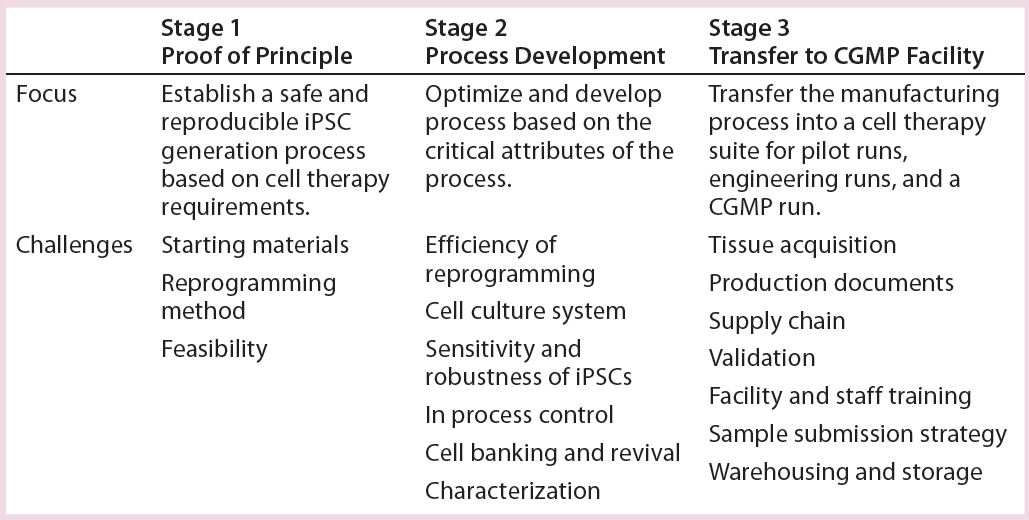
Table 1: A step-by-step approach leads to establishment of a robust, reproducible, CGMP- compliant iPSC manufacturing process. This is a three-stage approach: (1) establishing an iPSC generation process using a nonintegrating, episomal-based technology; (2) process optimization and protocol development based on critical attributes of the process; (3) transferring the manufacturing process into a CGMP manufacturing facility. A number of process design inputs were considered at each stage: choice of starting materials, safety of the reprogramming method, and feasibility in stage 1; efficiency of reprogramming, cell culture system for generation and expansion of iPSCs, their sensitivity and robustness, establishing in-process controls, cell banking and revival, and characterization (stage 2); and tissue acquisition, production documents, validation, sample submission strategies, and warehousing and storage (stage 3).
Stage 1: Proof-of-Principle Experiments
First, we focused on the starting material for iPSC derivation and safety of the iPSC generation method. Based on several practical and technical issues concerning tissue acquisition and hiPSC generation, we used CD34+ cells derived from cord blood as our starting material (5). Factors considered in making this decision included the availability of well-characterized cord- blood units with appropriate consent, widespread use of cord-blood units for clinical application, established protocols for collection and banking, and relatively efficient and fast generation of hiPSCs from cord- blood–derived CD34+ cells (compared with other starting cell types).
Reprogramming: Given the safety concerns surrounding the use of viral vectors initially used by Yamanaka and colleagues in 2007 for generation of clinical grade iPSCs (2), we selected a nonintegrating, episomal-based technology for our reprogramming process. This technology was initially developed by Cheng and colleagues based on the single transfection of two plasmids (6): pEB-C5 and pEB-Tg. The former is a polycistronic episomal reprogramming plasmid containing five genes encoding the transcription factors Oct4, Sox2, Klf4, c-Myc, and Lin28; the latter is an episomal plasmid expressing SV40 Large-T antigen demonstrated to increase reprogramming efficiency.
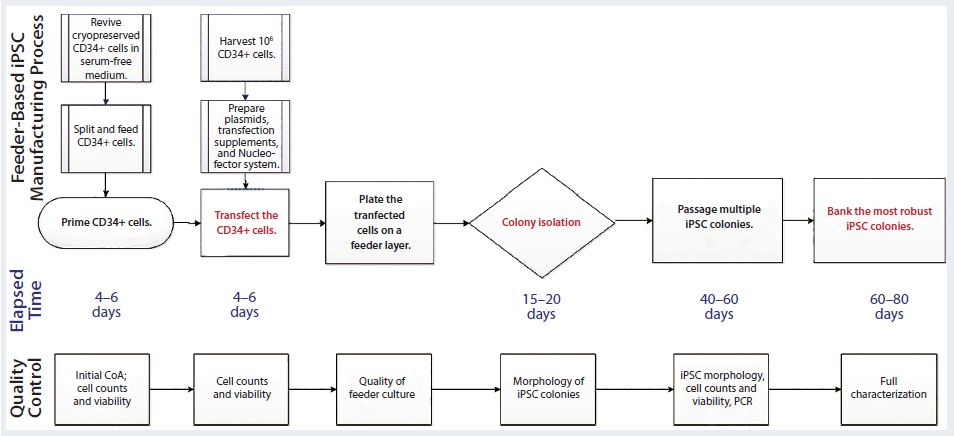
Figure 1: Human iPSC manufacturing uses the episomal-based reprogramming method in a mouse embryonic feeder-based cell culture system; overview shows the iPSC manufacturing process on a feeder-dependent system with processing time (days) and quality control activities including in-process control and final characterization. The manufacturing process includes a priming step for CD34+ cells, reprogramming and nucleofection of CD34+ cells, plating those cells on the feeder system, colony selection, passaging, expansion, and banking of iPSCs.
Figure 1 illustrates the iPSC manufacturing process using this episomal-based reprogramming method in a cell culture system based on mouse embryonic feeder (MEF) cells (6). Well-characterized, cryopreserved CD34+ cells (>80% purity) were used as the starting materials for this iPSC generation process. The cells were isolated from a cord-blood unit using a proprietary process designed for enriching CD34+ cells in an open-system column of magnetic beads. Cryopreserved CD34+ cells were thawed for a priming period of four to six days in a medium supporting their expansion. That priming step increased the purity and yield of CD34+ cells for reprogramming.
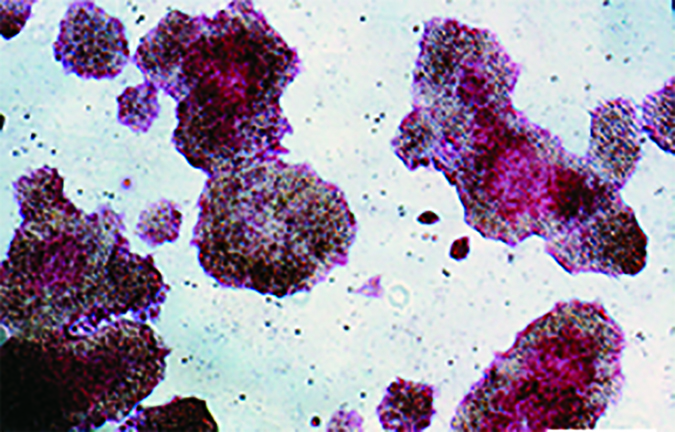
Following generation of iPSCs using a combination of pEB-C5 and pEB-Tg plasmids (using the Lonza Nucleofector 2 cuvette-based transfection system), multiple iPSC colonies (six to 10, minimum) were selected and serially subcultured on tissue-culture plates in a feeder-based culture system. Process design at this stage exhibited the reproducible generation of high-quality iPSCs using CD34+ cells isolated from multiple donors (10 different people) using episomal plasmids. Those were cleared from the iPSCs at passage levels 8–10 as evaluated by a semiqualified polymerase chain reaction (PCR) method based on gel electrophoresis (Figure 2).
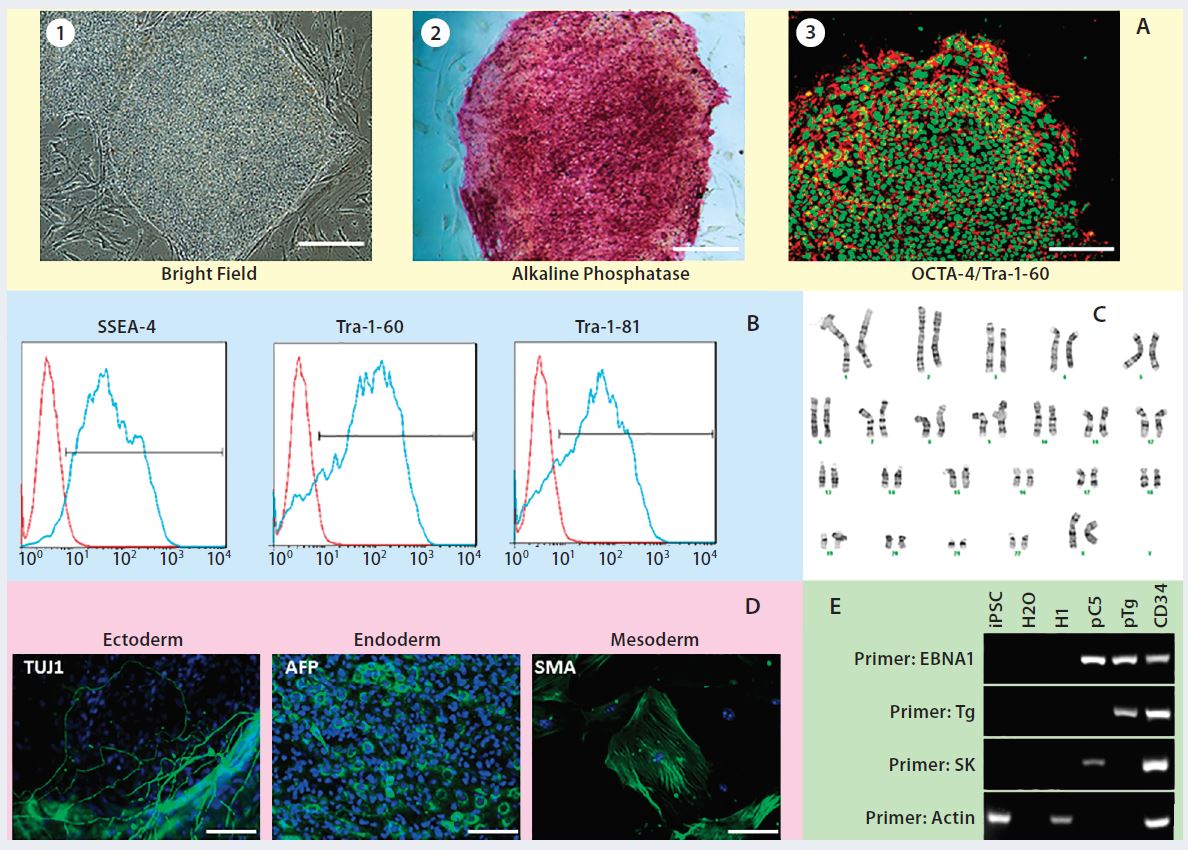
Figure 2: Human iPSCs generated using a feeder-dependent system during proof-of-principle stage (stage 1); (a) an iPSC colony positively stained with alkaline phosphatase (AP) and iPSCs expressing pluripotency markers OCT4 and TRA-1-60 (b) iPSCs expressing the pluripotent stem cell surface markers SSEA4, TRA-1-60, and TRA-1-81 (blue) with an isotype control (red); (c) iPSCs demonstrated normal karyotype after 10 passages; (d) iPSCs differentiated into embryoid bodies and readily expressing the markers for early ectoderm (TUJ1), endoderm (alpha-feto protein, AFP), and mesoderm (smooth muscle actin, SMA); (e) semiqualified PCR analysis of the iPSC sample exhibited plasmid clearance at P6. The top row shows plasmid (if applicable) or genomic DNA purified from different samples: iPSCs generated on feeders, water, human embryonic stem cells (H1), pEB-C5 plasmid, pEB-Tg plasmid, and CD34+ cells four days after nucleofection with pEB-C5 and pEB-Tg plasmids. Vertical column from top to bottom shows primers for EBNA1 vector, pEB-Tg, pEB- C5 (SK), and reference (actin). Scale bar in all images is 250 µm, except for (d) 125 µm.
Stage 2: Process Development
In the process development stage, we mainly focused on eliminating the feeder-based culture system for generation and expansion of iPSCs to address concerns around the use of the feeder system. We made efforts to improve the efficiency of our reprogramming process by incorporating enhancers to improve its robustness and address sensitivity issues with iPSCs growing in feeder- free cell culture systems.
Pluripotent stem cells (PSCs) are anchorage dependent and grow in the form of intact colonies of cells with a defined edge. They are susceptible to spontaneous differentiation without the necessary cell culture requirements to supress that phenomenon. The most critical cell culture requirements are an adequate substrate on which PCSs can attach and grow, a rich culture medium to support their proliferation while preventing spontaneous differentiation, and a passaging method to serially subculture the cells while maintaining high viability and pluripotency.
Process development was important although growth and expansion of iPSCs on MEF is robust. Such cells are sensitive to the type of surface matrix on which they can attach and to their growth medium. Both morphology and phenotypic characteristics can be altered by differences in either.
First, we modified the iPSC generation protocol described above by replacing the Lonza Nucelofector 2 system with a CGMP-compliant 4D-Nucleofector system and P3 solution kit (both from Lonza). We also evaluated the effects of different small molecules (sodium butyrate, TGF-beta inhibitor, and vitamin C) as potential enhancers for reprogramming efficiency, different physiological oxygen concentrations (hypoxia and normoxia), and different cell culture substrates. The latter included Synthemax and Matrigel substrates from Corning, StemAdhere substrate from Primorigen Biosciences, CELLstart substrate from Thermo Fisher Scientific, and Lonza’s L7 hPSC matrix. We tested each substrate using the growth medium recommended by its manufacturer (unpublished data).
Culture Medium and Passaging Solution: More important is that we developed a new cell growth medium: L7 hPSC medium. After preliminary evaluation, we determined that existing media (although effective at a laboratory scale) could not support reproducible generation of large numbers of iPSCs (>10 iPSC colonies) from different donors using the episomal plasmid technology (unpublished data). The new medium is proprietary, and its development and features are discussed in detail in a manuscript that is currently in preparation. It is a combination of L7 hPSC basal medium and L7 hPSC supplements. The basal medium is xeno-free and was specifically designed for maintenance and expansion of human PSCs by screening hundreds of basal medium supplements. The supplements contain xeno components that are commonly used in clinical manufacturing applications. However, an available xeno-free version of this proprietary medium is currently restricted to research use and can be used for every- other-day feeding of PSCs.
In evaluation of the different substrates, we found that the L7 hPSC matrix with the L7 hPSC medium and reprogramming enhancers and hypoxia conditions (3–5% O2) consistently generated human iPSCs from more than three donors under feeder-independent conditions. However, the efficiency of reprogramming was significantly lower than that under feeder- dependent conditions. This was a major challenge because our goal was to develop a reproducible manufacturing process that can produce high-quality iPSCs from CGMP-grade starting materials under stringent CGMP requirements. So we incorporated an Alhydrogel aluminium hydroxide (alum adjuvant) wet-gel suspension (Brenntag Nordic Biosector A/S), which significantly enhanced integration-free reprogramming under defined and feeder-free conditions (7).
We used a chemically defined, sodium-citrate–based, nonenzymatic L7 hPSC passaging solution for serial subculture of iPSCs (8). Using that solution, we serially subcultured the iPSCs generated under feeder-free conditions for about 30 passages (an average of five to six days each) in the form of cell colonies under feeder-free conditions in L7 hPSC medium on the L7 hPSC matrix.
Testing and Controls: To ensure that iPSCs generated using this reprogramming culture system could maintain normal karyotype following serial passaging, we conducted an important study to address safety concerns over long-term expansion of iPSCs. This study is designed for karyotype analysis of iPSCs generated under feeder-free conditions.
Briefly, CD34+ cells isolated from three different donors underwent the reprogramming process using a combination of episomal plasmids (pEB-C5 and pEB-Tg) and enhancers in the L7 reprogramming and cell culture system. Six iPSC colonies from each starting material were isolated at day 15, and then three iPSC colonies were maintained in the culture after passage 5 until passage 30. Complying with good laboratory practice (GLP), Cell Line Genetics, Inc., performed human G-banding karyotyping on nine iPSC colonies (three donors, three colonies each) at passage levels 5, 15, and 30. Results of this study demonstrated that all nine colonies maintained normal karyotype in the L7 cell culture system (Figure 3). In addition to this study, we conducted karyotype analysis every 10 passages as part of a routine characterization of iPSCs generated during the manufacturing process.
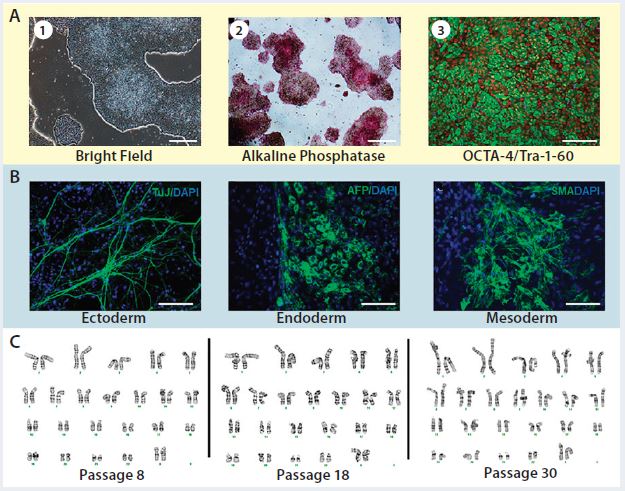
Figure 3: Karyotype analysis at different passage levels for human iPSCs generated using the Lonza CGMP-compatible, feeder-independent cell culture system; (a) iPSCs generated on the feeder-free system, some positively stained with alkaline phosphatase (AP), and some expressing pluripotency markers OCT4 and TRA-1-60; (b) iPSCs generated in the feeder-free system and differentiated into embryoid bodies, readily expressing markers for early ectoderm (TUJ1), endoderm (AFP), and mesoderm (SMA); (c) karyotype analysis of the same iPSC cell line generated under a CGMP- compliant process and serially subcultured at different passages — P8, P18, and P30. Scale bar in all images is 125 µm, except for (a) left and middle images, 500 µm.
As part of process development, we established appropriate in-process controls as well as tests for final- product characterization. In-process control studies focused on the quality of iPSCs generated under the feeder- free system and the processing point to cryopreserve, discard, or continue expanding iPSCs. It was important to ensure that only a limited number of high-quality iPSCs (identified by actively growing colonies with defined, smooth edges and little or no spontaneous differentiation) were maintained in the cell culture. In addition, we used a qualified quantitative PCR (qPCR) method as part of our in-process control strategy to evaluate the quality of each iPSC colony based on the level of residual plasmid clearance at different passages.
Characterization studies focused on establishing a product-release strategy based on
- iPSC identity evaluated by short tandem repeat (STR) and expression of pluripotency surface markers
- safety evaluated by sterility, endotoxin, mycoplasma, viral testing, and karyotype
- purity by testing the expression levels of pluripotency surface markers and STRs
- viability evaluated by cell count and viability measurements.
We considered additional tests such as embryoid body (EB) formation and immune-staining of iPSCs for information-only characterization (7).
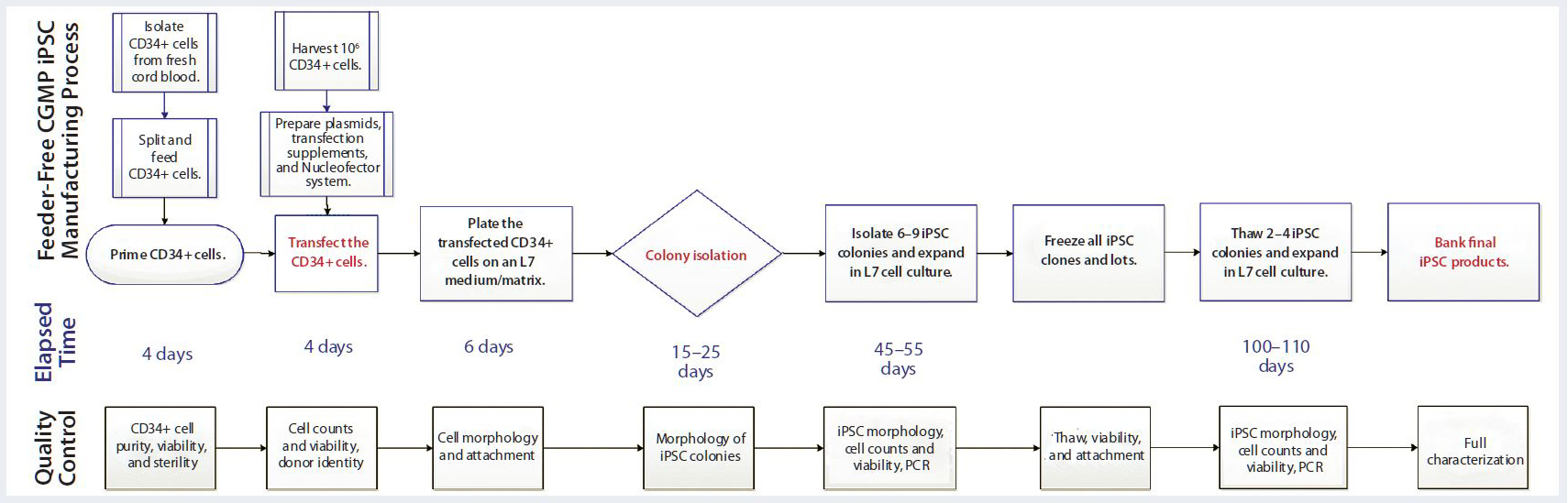
Figure 4: Overview of Lonza’s CGMP-compliant human iPSC manufacturing process based on a defined, feeder-free reprogramming and cell culture system, optimized at the end of stage 2, with processing time (days) and associated QC activities. The process includes a priming step for CD34+ cells, reprogramming and nucleofection of CD34+ cells, plating them on an L7 feeder-free cell culture system, colony selection, passaging, expansion, and banking of iPSCs using the L7 cell culture system. Quality control activities include characterization of starting CD34+ cells, intermediate iPSCs (based on quality and plasmid clearance), and final characterizations based on established testing and criteria.
Figure 4 is a schematic of the final CGMP-compliant iPSC manufacturing process. Compared with the initial process established for a feeder-dependent iPSC-generation process (Figure 1), this process took about 40 days longer but is designed to maintain a set process-design output. Priming and subsequent nucleofection of CD34+ cells is complete on day 4 (based on evaluation of CD34+ cell purity and growth) instead of the four to six days used to execute nucleofection in the feeder-dependent process.
Most important, we evaluated our CGMP-compliant process based on established in-process controls and qualified characterization assays. For example, we used a semiqualified (gel- based) PCR method to evaluate clearance of residual plasmids after reprogramming and expansion of iPSCs using the feeder-dependent process. By contrast, we used a quantitative PCR method to evaluate plasmid clearance under CGMP- compliant conditions. That method was qualified based on standard guidelines and a set of accuracy, specificity, limit of detection (LoD), and limit of quantification (LoQ) criteria (7).
Stage 3: Transfer of the Process
Following establishment of a robust iPSC manufacturing process with defined and reproducible process outputs, we focused on transferring that process into a CGMP facility. In moving from training runs into engineering runs and eventually CGMP manufacturing runs, we addressed a number of manufacturing challenges, including tissue acquisition, production documentation, supply chain, process validation, facility preparation, staff training, sample submission strategy, warehousing, and storage. Note that although those activities were reevaluated and finalized during stage 3, some (relating to tissue acquisition and supply chain, for example) had been initiated at earlier stages and/or in parallel to the process optimization activities in stage 2.
In addition, we initiated a parallel gap analysis and rectification process to identify deficiencies in characterization of starting materials, intermediates, and final product to minimize risks to CGMP manufacturing. We provide details of the studies and steps taken to transfer iPSC manufacturing from process development to CGMP facilities in a review accepted by Stem Cell Reports (7). Here we briefly summarize these activities.
Before CGMP manufacturing, the optimized iPSC manufacturing process was carried out multiple times within the scope of two training and two engineering runs. During the training runs, cell therapy technicians were trained and the first production-document drafts were developed, including master batch records (MBRs) and standard operating procedures (SOPs) for both the manufacturing process and bioassay services. During the engineering or validation runs, the final iPSC manufacturing process was reevaluated in the CGMP manufacturing facility using approved (or nearly approved) production documents. Execution of these process runs was critical to successful completion of the iPSC CGMP manufacturing process because they demonstrated that the process was ready to be executed in a CGMP manufacturing facility using standard documentation approved by our quality assurance (QA) department.
Some activities carried out during the transfer stage (e.g., instrumentation validation and CGMP facility preparation) were performed based on standard CGMP manufacturing guidelines that are not specific to particular products. By comparison, other activities such as tissue acquisition and supply chain management are considered iPSC-specific process requirements for compliance with quality standards.
In particular, our tissue-acquisition program for CGMP manufacturing of iPSCs was based on 21 CFR 1271 guidelines to procure CGMP-grade fresh cord- blood units (8). The most critical tissue-sourcing activities included defining tissue requirements, working with a tissue-recovery agency, establishing forms and SOPs, and recovering tissue and donor eligibility determination. Supply chain activities associated with CGMP manufacturing of iPSCs mainly focused on establishing a complete list of raw materials intended for the manufacturing process as well as relevant QC and bioassay tests, vendor qualifications, raw-material part set-ups, and inventory management (storage conditions, expiration tracking, reorder points, and material flows to and within GMP manufacturing facilities).
After reevaluating our process through the multiple training and engineering runs, we carried out a CGMP manufacturing run using CGMP-grade fresh cord-blood unit with approved donor-eligibility criteria. CD34+ cells were isolated using a CliniMACS CD34 reagent system from Miltenyi Biotec. Then cells were introduced into the priming stage, in which their purity increased from 89.10% on the day they were isolated to 94.9% four days later, exhibiting a greater than threefold expansion.
Multiple iPSCs were generated during reprogramming, but two clones were maintained in culture after in-process evaluation and an intermediate cryopreservation step. Evaluation of both clones/lots using the qualified PCR method revealed that both lots were clear of residual plasmids, and >150–200 vials (2 × 106 cells/vial) were cryopreserved at the end of the process.
In QC analysis of the CGMP manufactured iPSCs, the cells met the predefined characteristics of pluripotent stem cells, including recovery after thawing, expression of pluripotency markers (Oct4, Nanog, Tra1-60, Tra1-81, and SSEA4) tested by flow cytometry, EB formation, genetic identity matching with starting material tested by STR analysis, normal karyotyping, and other standard safety assays such as tests of sterility, mycoplasma, viral safety, and endotoxins (7).
Results confirm that our iPSC manufacturing process is robust and reproducible and can be carried out under CGMP guidelines to generate high-quality iPSCs for manufacturing of clinical-grade specialized cells for cell-restorative transplantation strategies. We have shown that iPSCs generated in each stage of our process (including process development, training and engineering runs, and a GMP manufacturing run) meet pluripotent stem-cell characteristics as evaluated by standard and qualified assays. Lonza’s CGMP manufacturing process for iPSCs has been submitted to the US Food and Drug Administration through a drug master file (DMF number BBMF-16567), which can be accessed by reference when filing an investigational new drug (IND) application.
References
1 Thomson JA, et al. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282, 1998: 1145–1147.
2 Takahashi K, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts By Defined Factors. Cell 131, 2007: 861–872.
3 Chen G, et al. Chemically Defined Conditions for Human iPSC Derivation and Culture. Nat. Meth. 8, 2011: 424–429.
4 Dowey SN, et al. Generation of Integration-Free Human Induced Pluripotent Stem Cells from Postnatal Blood Mononuclear Cells By Plasmid Vector Expression. Nat. Protoc. 7, 2012: 2013–2021.
5 Rao M, Ahrlund-Richter L, Kaufman DS. Concise Review: Cord Blood Banking, Transplantation and Induced Pluripotent Stem Cell: Success and Opportunities. Stem Cells 30, 2012: 55–60.
6 Chou BK, et al. Efficient Human iPS Cell Derivation By a Non-Integrating Plasmid from Blood Cells with Unique Epigenetic and Gene Expression Signatures. Cell Res. 21, 2011: 518–529.
7 Baghbaderani et al. CGMP Manufactured Human Induced Pluripotent Stem Cells Are Available for Pre-clinical and Clinical Applications. Stem Cell Rep. 2015: in press.
8 CBER. Guidance for Industry: Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/P) — Small Entity Compliance Guide. Code of Federal Regulations Title 21, Part 1271. US Food and Drug Administration: Rockville, MD, August 2007.
Behnam Ahmadian Baghbaderani is a senior scientific program director, and Thomas Fellner (thomas.fellner@lonza. com) is director of cell therapy commercial development at Lonza Pharma and Biotech, 8830 Biggs Ford Road, Walkersville, MD. Mahendra Rao is founder and chief scientific officer of Mahendra Rao, LLC. L7 and Nucleofector are registered trademarks of Lonza Pharma and Biotech.