Cell, gene, and tissue (CGT) therapies and other advanced-therapy medicinal products (ATMPs) have made tremendous progress over the past decade. They are different from other biologics and small molecules because of their inherent complexity and variability. Although many unknowns remain about the development of these products, their clinical success has enabled the CGT therapy and ATMP fields to advance rapidly. We are seeing an increase in the number of marketing authorization applications (MAAs) filed in the European Union and new drug applications (NDAs) filed in the United States for these and other products in the pipeline. The US Food and Drug Administration (FDA) and European Medicines Agency (EMA) expect an upsurge in the number of CGT therapies and ATMPs to be launched on the market in the near term (1, 2).
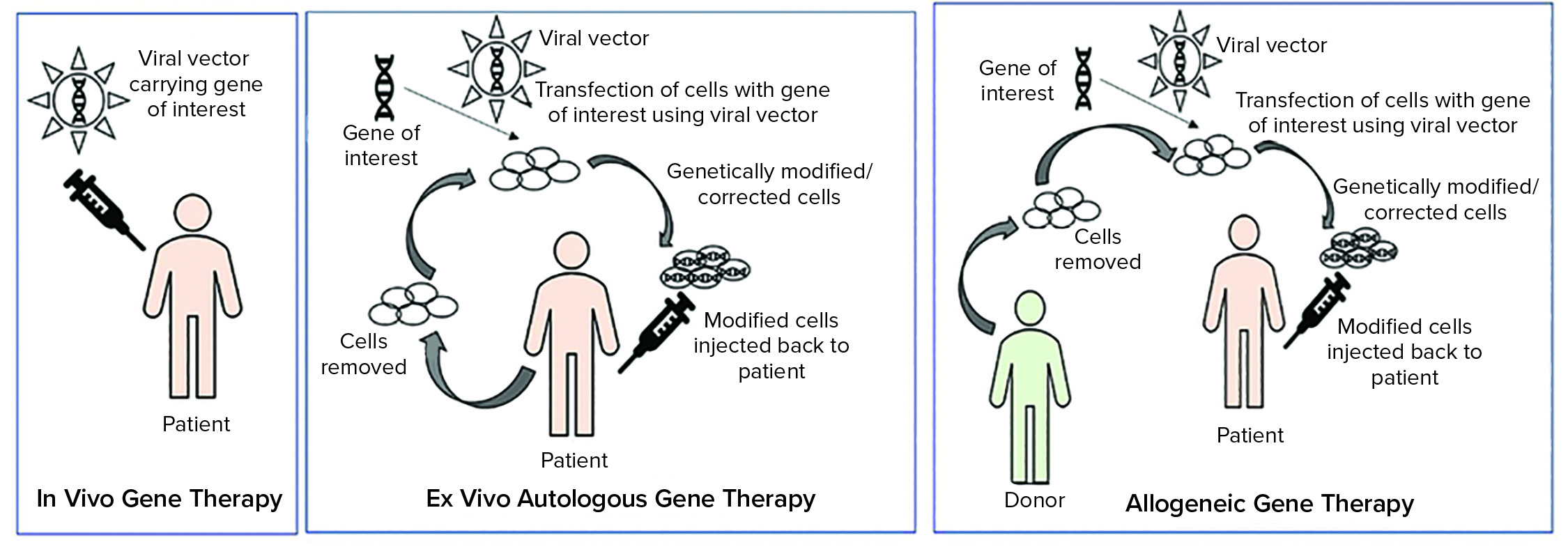
Figure 1: Types of gene therapies
Development of such advanced products involves tight timelines and high costs, and several biomanufacturers have complex but novel approaches to nonclinical and clinical programs. Product quality related to chemistry, manufacturing, and controls (CMC) also must be maintained. Part 1 of this article highlights challenges encountered during the manufacture of CGT therapies and ATMPs.
Development of Cell, Gene, and Tissue Therapies
In the United States, cellular and gene therapy products include human cells, tissues, and cellular and tissue-based products (HCT/Ps). Referred to as ATMPs in Europe, they currently constitute one of the most advanced and rapidly growing fields of medicinal treatment. The EMA classifies ATMPs into gene therapy, somatic cell therapy, or tissue-engineered products in accordance with Article 17 of Regulation (EC) 1394/2007. The FDA regulates HCT/Ps under 21 CFR part 1271 Human Cells, Tissues, and Cellular and Tissue-Based Products in conjunction with the appropriate section of the Public Health Service (PHS) Act (3). Sections 361 and 351 relate to the level of risk that a biological product developed as a medicinal product for human use potentially poses to patients. Section 361 applies to products deemed to be of lower risk. Section 351 clearly identifies and classifies products that are more than minimally manipulated and not intended for homologous use, making them subject to greater regulation under 21 CFR 1271 Subpart D Good Tissue Practices (see “EMA and FDA Definitions” box).
Tissue and cellular ATMPs can be autologous (tissues or cells that are obtained for the starting material are from the patient) or allogeneic (tissue or cells that are obtained from starting material from a different donor) (5).
As Figure 1 illustrates, ex vivo autologous gene therapy involves extraction of cells from a patient (e.g. chimeric antigen receptor (CAR) T-cell therapy through leukapheresis). Those cells are modified with a gene of interest by using a viral vector, and genetically modified or corrected cells are injected back into the patient. In vivo gene therapy involves injecting a vector (usually a viral vector) carrying a gene of interest to a patient.
Development of gene therapies is complex, so their CMC can be difficult at different manufacturing steps. Appropriate controls must be in place to generate an acceptable quality of drug product, despite a number of issues such as bespoke and complex materials and equipment, multiple banking requirements (mostly in case of gene therapies or allogeneic cell therapies), nonuniform quality of ancillary starting materials, novel manufacturing processes, sterility requirements, adventitious agent risks, difficult-to-measure in-process controls (IPCs), complex analytical methods, low material availability, and limited shelf life.
As discussed below, an adequately controlled system for raw materials, starting (source) materials, and reagents is the foundation for manufacturing. As the manufacturing of a product transitions from small- to large-scale (or in case of autologous cell therapies, to multiple scales), environmental control and site monitoring are required along with an increasing dependence on automation to improve robustness and quality of CGT therapies and ATMPs. However, technology transfer can be problematic when such products are transferred from bench to good manufacturing practice (GMP) scale.
| EMA and FDA Definitions |
| Acccording to Article 2 of Regulation (EC) No. 1394/2007-26, the EMA defines the following terms.
A gene therapy medicinal product is a biological medicinal product that contains a recombinant nucleic acid used in/administered to human beings with a view to regulating, repairing, replacing, adding, or deleting a genetic sequence. Its therapeutic, prophylactic, or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence. A somatic cell therapy medicinal product is a biological medicinal product containing cells or tissues that have been subject to substantial manipulation so that biological characteristics, physiological functions, or structural properties relevant for the intended clinical use have been altered, or of cells or tissues that are not intended to be used for the same essential function(s) in the recipient and the donor. It is presented as having properties for or is used in/administered to human beings with a view to treating, preventing, or diagnosing a disease through the pharmacological, immunological, or metabolic action of its cells or tissues. A tissue-engineered product contains engineered cells or tissues. It is presented as having properties for or is used in/administered to human beings with a view to regenerating, repairing, or replacing a human tissue. The FDA defines gene therapy as a technique that modifies a person’s genes to treat or cure disease. Gene therapy products are defined as follows (4). Plasmid DNA is “a circular DNA molecule that can be genetically engineered to carry therapeutic genes into human cells.” Viral vectors are “used to carry therapeutic genes into human cells. Viruses have a natural ability to deliver genetic material into cells. Thus, some gene therapy products are derived from viruses. Once viruses have been modified to remove their ability to cause infectious disease, these modified viruses can be used as vectors (vehicles).” Bacterial vectors “can be used to carry genes into human tissues. Bacteria can be modified to prevent them from causing infectious disease and then used as vectors (vehicles).” Human gene-editing technology is “a strategy of editing genes with the goal of disrupting harmful genes or to repair mutated genes.” Patient-derived cellular gene therapy products involve removing cells from a patient, genetically modifying those cells (often using a viral vector), and then returning them to the same patient. |
Manufacturing Challenges
Control of Raw Materials, Starting Materials, and Reagents: The quality of starting and raw materials is a key factor in ATMP production. Biomanufacturers are responsible for the quality of their materials sourced for production. Raw materials, starting materials, and reagents should be GMP-sourced and take into consideration Ph.Eur. 5.2.12 Raw Materials of Biological Origin for the Production of Cell-Based and Gene Therapy Medicinal Products (6). In the United States, developers of HCT/Ps are encouraged to adopt a risk-based approach to assess the quality of raw and ancillary materials used in accordance with USP chapter <1043> “Ancillary Materials for Cell, Gene, and Tissue Engineered Products” (7). That item often is a sticking point particularly with the manufacturing of important starting materials such as viruses (e.g., gamma retroviruses and lentiviruses) used as vectors in gene-modified immune cells (e.g., CAR T cells critical for the overall safety and efficacy of a final product).
A risk-based approach also is emphasized for viruses such as adenoassociated viruses (AAV) that are developed and marketed as in vivo gene therapies. Detailed descriptions and evidence of viral safety and testing of raw and ancillary materials should be provided in regulatory dossiers early in product development and should be in place by the time of first-in-human (FiH) clinical studies. That documentation provides assurance that raw and ancillary materials are free from communicable agents such as adventitious agents, viruses, and transmissible spongiform encephalopathies (TSE) (e.g., bovine spongiform encephalopathy, BSE). In some cases, it is sufficient for developers to provide regulators with evidence that the highest available quality (with no risk to safety) is being used. However, that common scenario for CGT therapies and ATMPs should be discussed upfront with regulators early in a development program to prevent delays later on, especially with processes that use only research-grade materials.
Quality agreements should be in place between suppliers and manufacturers to ensure that starting materials such as plasmids, vectors, cell banks, and specific or novel reagents are of acceptable grades. Suitable tracking and traceability systems also should be parts of those agreements. They help prevent deviations in material quality. But if deviations still occur, proper steps can be taken to replace either the materials or the suppliers.
ATMPs were developed first as autologous products, and most tissue-engineered products remain as such. However, from a CMC and marketing perspective, allogeneic products are preferable. They enable the manufacture of “off-the-shelf” products and reduce the bespoke-manufacturing cost burden. In general, sourcing material from suitably screened healthy donors rather than patients improves a product’s safety profile and provides material of greater consistency. By contrast, the quality of starting material obtained from a patient depends on that person’s health, age, and illness, among other factors. The procurement, processing, donor eligibility determination, screening, and testing of allogeneic starting materials such as apheresis blood products or mesenchymal stem cells from bone marrow are governed in Europe by the EU Blood Directive 2002/98/EC 27 and the EU Cells and Tissues Directive 2004/23/EC, respectively (8) and in the United States by 21 CFR 1271 Subpart C Donor Eligibility (9, 10). As discussed in those directives and adjacent frameworks, multiple communicable agents are required to be absent from donor starting material within the appropriate testing period. Such agents include human immunodeficiency virus types 1 and 2, human T-cell leukemia virus types 1 and 2, hepatitis B virus (HBV), hepatitis C virus (HCV), Chlamydia trachomatis, Neisseria gonorrhoea, Treponema pallidum (syphilis), West Nile virus (WNV), and Zika virus. Developers must be aware that such requirements can depend on jurisdiction and region. For example, there is a marked difference in the expectation of the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan with regard to the list of viruses of concern and testing periods compared with those given by the EMA and the FDA.
Drug developers must ensure that blood- and tissue-collection facilities have appropriate regional registration and accreditation. The stability of starting materials collected after sourcing must be understood, and appropriate controls must be in place for their transport and storage to manufacturing sites. Even early in development, critical quality attributes (CQAs) should be understood and defined as well as possible.
Release and stability testing for autologous products can be a challenge because of the bespoke nature of their manufacture. Compared with autologous therapies, allogeneic products more closely resemble traditional biologics in their development with respect to characterization, specifications, release, and stability testing before administration.
Theoretically, manufacturing experience of allogeneic products is gained more readily through the production of multiple drug substance batches and drug product lots. So regulators expect evidence of complete and comprehensive validations of processes with representative process performance qualification (PPQ) batches, which are more akin to those for biologics. Autologous products are manufactured based on a “vein-to-vein” model: Starting material is isolated from a patient. After a manufacturing process, genetically modified product is administered directly back to the same patient. Those general aspects are the same with other cell therapies. However, autologous tissue-engineered products often require a surgical procedure for administration as well.
By their nature, starting materials taken from patients are highly variable, thus exerting considerable pressure on manufacturing processes and identification both of product CQAs and critical process parameters (CPPs) needed to control them. Such pressures can hinder efforts to characterize and establish specifications for acceptance criteria used for release testing. That increases the likelihood of out-of-specification results during release testing of CGT drug products.
Typically, manufacturing autologous gene-modified immune cells (e.g., CAR-T and natural killer, NK, cells) takes two to three weeks, including final release testing. Manufacturing generates a single drug-product lot, which typically is not enough to support a stability program. Once a process has been initiated and leukapheresis material has been isolated from an already weak and vulnerable patient, it is ethically important that a product be manufactured, released, and administered to that patient without delay — even if the product can be frozen after manufacture. Such a short timeline does not account for pharmacopeial testing for sterility, which can take 14 days. Manufacturing a tissue-engineered product can take even longer. Sterility testing usually requires several steps, and product release is based only on preliminary data because such products cannot be frozen.
For all CGT products and ATMPs, rapid sterility and mycoplasma testing using polymerase chain reaction (PCR) strategies and chemiluminescence methodologies have received regulatory acceptance when adequate qualification and validation for specificity, sensitivity, and robustness have been demonstrated (11).
Comparability: During all phases of development and after approval, CMC changes might be required. Such changes could be made to manufacturing components and materials, process steps, and facilities, for example. To confirm that such pre- and postchanges do not affect the CQAs of a product significantly, a comparability assessment must be completed.
That type of assessment will be simpler in early development stages and more comprehensive as developers move through later stages of development. The best approach is to use a development-stage–specific risk-based strategy. Establishment of CQA comparability enables developers to leverage clinical data generated using prechange material moving forward. If manufacturing process changes must be made during development and comparability cannot be demonstrated, then clinical studies might need to be repeated using material generated from the current (and the intended commercially representative) process.
Automation and Environmental Control: Development of many ATMPs begins in academic or research institutions attached to hospitals, where production takes place at small scales. Due consideration should be given to improving manufacturing processes and to networking with potential investors for successful commercialization of those products. Some institutions lack the industry and regulatory expertise and facilities to transition into commercial manufacturing (especially considering the complex nature of ATMPs). Early involvement with relevant experts and regulatory agencies can improve understanding of the requirements for scaling up to a commercial-scale GMP environment.
However, some hospitals and academic institutions have developed on-site GMP bioprocessing facilities and are fully capable of supporting preclinical to phase 2/3 trials. Some large pharmaceutical and biotechnology companies rely on those institutions for clinical trials and use in-house facilities or contract manufacturing organizations (CMOs) for commercial supply.
Cleanroom requirements are important to consider for manufacturing CGT therapies and ATMPs to prevent cross-contamination with other products. In the United States, such requirements are ISO 5 with ISO 7 background. Corresponding requirements in the European Union are grade A areas with grade B background or grade A in grade C (or even grade D) backgrounds if closed systems are used (12). CMOs have multiple clients, each with distinct processes, and those processes need cleanrooms. With the large number of ATMPs under development and the limited number of CMOs undertaking the manufacturing of such products, cleanroom availability has been a burden. Associated manufacturing costs, especially if contamination occurs, would be significant. Hence, the ATMP industry is moving toward automated and fully closed systems. Such “plug and play’’ systems enable small-scale manufacturing typical for an autologous therapy as well as parallel processing of other products at the same CMO facility with low risks of cross-contamination.
CMOs also have started investing in capabilities to add cleanroom boxes to their facility to increase capacity. Such facilities need to be GMP compliant, and the “GMP for ATMPs” guidance (12) provides some flexibility for ATMPs. Third-party organizations such as the Foundation for the Accreditation of Cellular Therapy (FACT) and the Joint Accreditation Committee of the ISCT, Europe (JACIE) also have produced standards to ensure supply of high-quality cell and gene therapy products (13). Those standards cover all activities from collection to administration. Although not mandatory, certifications from such organizations are valuable because they are aligned with both US and EU requirements and provide proof that a manufacturing facility is GMP compliant and has an appropriate quality management system (QMS).
Technology Transfer Challenges
Technology transfer for small molecules and traditional biopharmaceuticals is far more streamlined than it is for ATMPs because complex activities can vary from one product to another. Sufficient time and effort should be spent on due diligence to determine whether the company to which technology is being transferred meets the needs of the product sponsor. That forms the core of successful manufacture and delivery of a product. Key factors influencing technology transfer include a well-documented scope of activities and expectations, a multidisciplinary team with strong technical expertise, risk-analysis and risk-management programs, and achievable timelines and costs (14).
Open and transparent communication between both sites is critical. Other factors to consider are phase-appropriate and qualified analytical methods and acceptance criteria, well-defined training runs, engineering runs, process qualification (PQ), GMP (including cleanroom requirements) for each run, approved manufacturing batch records (MBRs), and standard operating procedures (SOPs). Sponsors typically use data from engineering and PQ runs for comparability assessments against specification criteria and to file investigational new drug (IND) applications and clinical trial applications (CTAs) (15).
Reduced CMC Timelines
Funding boards and stakeholders can pressure a manufacturer to reduce the timelines for IND application and CTA filings, and that can result in dramatically shortened CMC timelines. Industry experts advise biomanufacturers to find a balance between risks and accelerated CMC timelines by conducting appropriate risk-based studies. That would enable a streamlined approach and ensure that critical quality requirements are met. Depending on the program, biomanufacturers can provide data to regulators in a “rolling” approach during review or postapproval. Doing so could help instill flexibility in planning. Rushing through technology transfer activities without adequately establishing a process can lead to unexpected quality issues with clinical supply, which then can extend overall timelines and increase costs.
One commonly encountered issue in development of CGT products and ATMPs is the failure to launch or plan for launch. During development, timelines often do not allow sufficient attention to be given to postapproval supply logistics and CMC changes required to facilitate them. Thus, we often advise drug developers to consider those issues as part of development and before MAA and BLA submission. Using a postapproval change-management protocol for submission as part of MAA or BLA submission can aid in launch planning and enable sponsors to reach an early agreement with regulatory agencies regarding the approach to planned postapproval changes. Risk analysis should be consistent and proactive to identify unexpected risks, and a strategy should be put in place to manage those risks. These assessments should be live documents and updated routinely after key project milestones.
Compliance and Regulatory Affairs
CGT products and ATMPs have gathered interest among physicians and patients because of their ground-breaking therapeutic results and curative nature. To bring their products to market, developers should take advantage of all existing opportunities to keep abreast of successes and failures in this field. Upfront development activities and risk-based assessments are critical for reducing CMC risks during and after development. That and early involvement with regulators are the keys to success.
Part 2 will conclude this discussion with a focus on regulatory guidance for ATMPs, including the support and incentives offered by different regulatory agencies.
References
1 Bender E. Regulating the Gene Therapy Revolution. Nature 564(7735) 2018: S20–S22; https://doi.org/10.1038/d41586-018-07641-1.
2 Eder C, Wild C. Technology Forecast: Advanced Therapies in Late Clinical Research, EMA Approval or Clinical Application via Hospital Exemption. J. Mark. Access Health Policy 7(1) 2019: 1600939; doi:10.1080/20016689.2019.1600939.
3 21 CFR, Subchapter L: Regulations Under Certain Other Acts Administered by the Food and Drug Administration, Part 1271 Human Cells, Tissues, and Cellular and Tissue-Based Products. Fed. Reg. 66, January 2001: 5466; https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=1271.
4 What Is Gene Therapy? US Food and Drug Administration: Rockville, MD, 2018; https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy
5 The Glossary for Cell and Gene Therapy and Regenerative Medicine. Regen. Med. 13(8) 2018: S1; ; https://www.regmednet.com/the-glossary-for-cell-gene-therapy-and-regenerative-medicine-2018.
6 Pugieux-Amarantos C. General Chapter 5.2.12: Raw Materials of Biological Origin for the Production of Cell-Based and Gene Therapy Medicinal Products. European Pharmacopoeia Training Session on Biologicals, Strasbourg, France, 7–8 February 2017; https://www.edqm.eu/sites/default/files/cell_and_gene_therapy_by_celine_pugieux-amarantos-bio-training-feb2017.pdf.
7 <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products. USP29–NF24. US Pharmacopeial Convention: Bethesda, MD.
8 Commission Directive 2006/17/EC: Implementing Directive 2004/23/EC of the European Parliament and of the Council as Regards Certain Technical Requirements for the Donation, Procurement, and Testing of Human Tissues and Cells. Off. J. Eur. Union 8 February 2006; https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:038:0040:0052:EN:PDF.
9 Guidance for Industry: Eligibility Determination for Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products. US Food and Drug Administration: Rockville, MD, August 2007; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/eligibility-determination-donors-human-cells-tissues-and-cellular-and-tissue-based-products.
10 Testing Donors of Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/P): Specific Requirements. US Food and Drug Administration: Rockville, MD, 2019; https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/testing-donors-human-cells-tissues-and-cellular-and-tissue-based-products-hctp-specific-requirements.
11 Guidance for Industry: Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). US Food and Drug Administration: Silver Spring, MD, January 2020; https://www.fda.gov/media/113760/download.
12 Guidelines on Good Manufacturing Practice specific to Advanced: Therapy Medicinal Products. EudraLex: The Rules Governing Medicinal Products in the European Union, Volume 4 — Good Manufacturing Practice. European Commission: Brussels, Belgium, 22 November 2017; https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2017_11_22_guidelines_gmp_for_atmps.pdf.
13 Hourd P, et al. Regulatory Challenges for the Manufacture and Scale-Out of Autologous Cell Therapies. StemBook. Harvard Stem Cell Institute: Cambridge, MA, 2014; https://doi.org/10.3824/stembook.1.96.1.
14 Labant MA. Get Ready, Get Set, GMP and Cell Therapy: Considerations for Getting Started in Manufacturing with GMP. Gen. Eng. News 6 November 2018.
15 Manufacturing Matters in Cell Therapy. NSight NB Research Report. Nelsen Biomedical LLC: Eagan, MN, 2016.
Anjali Apte is manager of regulatory affairs, CMC (anjali.apte@pharmalex.com); Adeyemi Afuwape is associate director of regulatory affairs, CMC (adeyemi.afuwape@pharmalex.com); Zaklina Buljovcic is director of scientific services and principal consultant, innovative therapies (zaklina.buljovcic@pharmalex.com); and Zeb Younes is director regulatory affairs, CMC (zeb.younes@pharmalex.com), all at PharmaLex.