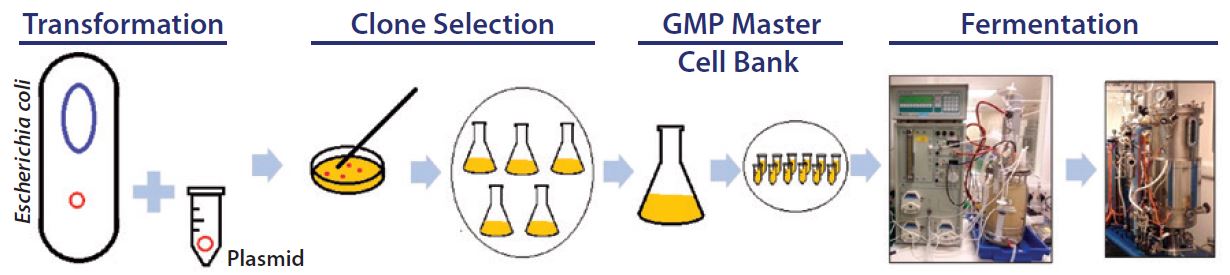
Figure 1: Development of production process for manufacturing plasmid DNA
DNA-based gene therapy products have been in clinical development since the 1990s. But over the past 24 months, the overall demand and therapeutic applications for plasmid DNA (pDNA) have rapidly grown and expanded. Currently, pDNA can be used directly as a therapeutic agent (e.g., in gene therapy or generation of vaccine antigens) and indirectly for a range of applications. Those include its use as a critical starting material for transient transfection to produce both viral-vector constructs (e.g., lentivirus or adenoassociated virus) and transient proteins in development (e.g., monoclonal antibody). Furthermore, pDNA is used as a master-template product to support production of new and emerging biopharmaceutical products and processes (e.g. RNA, in vitro protein production, and cell therapies).
Plasmids used as therapeutic agents tend to be relatively small in size (4.5– 6.0 kb and even down to 2.5 kb) (1), requiring relatively low quantities to support early phase clinical trials (1–5 g of purified pDNA). However, when pDNA is used as a transfection material, multiple plasmid constructs often are required to produce an individual product coupled with an escalating demand for plasmid quantity as production scales increase. In addition, pDNA sizes used for viral constructs are usually larger (≥11 kb) (1) and can contain base-pair sequences (such as inverted terminal repeats) that cause problems in their plasmid production.
Cobra Biologics has been producing plasmid DNA for a number of clinical applications for over 15 years at an approved site in the United Kingdom under the European Union (EU) clinical trials directive. The company has developed its own manufacturing process for production and purification of pDNA with related proprietary technologies. Those include an antibiotic-free plasmid maintenance system, operator repressor titration (ORT), and cell-lysis technologies (2–4). During this time, the company has successfully executed more than 240 DNA programs with process development, manufacturing, and stability studies for more than 35 different customers globally. Generated plasmids have been used in clinical trials for gene therapy and vaccines as well as in viral vector production.
Production of pDNA for clinical gene therapy and vaccine trials within the European Union must comply with current good manufacturing practices (CGMPs). Manufacturing standards for pDNA to be used as starting material in production of cell-based and gene-transfer medicinal products (GTMPs) are relatively undefined. Product sponsors are responsible for ensuring that materials are of suitable quality for their intended use. Points to consider are aligned with CGMP manufacturing requirements and include full traceability of raw materials entering a manufacturing process, elimination of components from human or animal origin, and development of a suitable testing strategy. Qualified cell banks should be used and tested for identity, viability, strain, genotype/phenotype, and presence/copy number of the plasmid vector of interest, along with a description of its structural elements. A pDNA product must be tested for sterility and purity levels (with removal of host-cell–derived proteins, residual host-cell DNA, RNA, and endotoxins to determined levels) and quantified for molecular isoforms (e.g., supercoiling, open circle). Identification by sequencing is a final requirement (5).
Plasmid DNA production is almost exclusively based on Escherichia coli Gram-negative bacteria. Production by E. coli is preferable for its relative simplicity, with rapid and inexpensive high-density cultivation, well-known genetics, and many compatible molecular tools available. E. coli also remains a valuable organism for high- level production of certain recombinant proteins.
Plasmid production typically involves E. coli K12 strains such as DH1, DH5, DH5a, JM109, JM108, and DH10B (6). K12 strains have been approved for a number of processes by the US Food and Drug Administration (FDA) and can be applied to recombinant protein and plasmid DNA production. In addition, all major national and international guidelines on biological safety for work with recombinant DNA technology have classified K12 as a biologically safe vehicle for propagation of many efficient gene- cloning and expression vectors (7).
To date, pDNA production processes have yielded 0.2–2.1 g plasmid per liter of culture media and productivities of 20–44 mg pDNA/g dry cell weight (DCW) using standard high-copy pUC origin-containing plasmids (7). The E. coli biomass containing pDNA typically is recovered either by centrifugation or tangential-flow filtration (TFF) followed by an alkaline or heat lysis to release the pDNA. Purification is achieved through a combination of ribonuclease treatment, selective precipitation, TFF, and chromatography. Those operations remove impurities such as open-circle, linear, multimeric, and irreversibly denatured forms of pDNA as well as host-cell genomic DNA, RNA, proteins, and endotoxins present in cell lysate. The FDA has set recommendations for acceptable levels of contaminants with pDNA at <1% w/w genomic DNA, RNA, and host- cell proteins (HCPs), <40 EU endotoxin/mg plasmid, in a solution that is >80% supercoiled pDNA (8). Those values are mainly guidelines, however, with normal industry expectations for supercoiled pDNA typically about >90% (1).
Development, Technology Transfer, and Production of pDNA: An efficient fermentation process is needed for producing large quantities of pDNA. Plasmid constructs are highly variable in size, backbone, and gene inserts, which leads to a broad range of cell culture productivity and highly variable levels of product and host-related contaminants that must be removed in purification (1).
The primary goal in designing a fermentation process for plasmids is to maximize both the volumetric yield (mg pDNA/L culture media) and specific yield (mg pDNA/g DCW or mg/L OD600nm) of supercoiled pDNA (sc-pDNA). It is also critical that fermentation produces high-quality sc-pDNA that minimizes process impurities that have similar properties to the sc-pDNA product, such as open- circle, linear, and nicked forms of pDNA that would be less effective therapeutically than the sc-pDNA form.
Development of a pDNA production process (Figure 1) starts with creation of a plasmid containing the gene of interest and then selection of a host strain to express it. The plasmid is transfected into the selected host strain, and after clonal selection and preparation of cell banks, small-scale production normally begins. At this point, optimization of culture conditions (media components, pH, temperature, and dissolved oxygen) and adequate induction strategies are established, generally through studies of biomass and plasmid yield and quality (6). High-yield pDNA production relies on synergy among host strain, plasmid, media, and induction strategy. Screening as many variables as quickly and cost- effectively as possible is the main goal during development.
Transfer from small-scale technology to manufacturing scale requires management of the scale-up process. Given the potentially sensitive and diverse response of cells to each transport phenomenon — influenced by impeller design, system geometry, scale, fluid properties, and operating parameters — this is not just a simple multiplication of relevant factors. Designing a large-scale pDNA production process requires not only a correct fermentation strategy to improve yields while reducing production costs, but it also requires skill and time investment while incurring costs (9, 10).
Application of Single-Use Technologies: A number of factors characterize the transition of an upstream process from either a small-scale bench-top bioreactor or traditional stainless steel vessel to a single-use system. Single-use technologies enable development of platform processes that rapidly produce a large range of plasmids for research and clinical application. In addition, disposables eliminate the need for cleaning, cleaning validation, and preparation of glass and steel containers and devices, enabling researchers and operators to focus on process-related tasks. Elimination of cleaning validation is particularly important for CGMP processes because of the low detection limits of assays commonly used to measure contaminants as well as a lack of acceptable cross-contamination levels for batches of different pDNA products.
A number of single-use technologies applicable to upstream processes are currently available with rocking and stirred-tank bioreactor systems (11). They were primarily developed for mammalian cell culture operations. Recently, alternative rocking reactor designs and innovative single-use stirred-tank bioreactor-type systems have become available. The new systems can meet the unique requirements of microbial cultures: e.g., increased demands of oxygen transfer capacity and tighter temperature controls.
Here, we evaluate alternative single-use systems for production of plasmid DNA material from E. coli cultures. We compared a TAP BioSystems single-use ambr250 modular system from Sartorius Stedim Biotech AG (specially tailored for microbial applications and targeted for high-throughput development and optimization activities) with a traditional 7-L bench-top glass bioreactor (Applikon Biotechnology). We also compared a Biostat CultiBag RM 20 rocking bioreactor system from Sartorius Stedim that used a 10-L CultiBag vessel. Suited to production of CGMP material for clinical trials, both the Biostat CultiBag RM 50 bioreactor and an Xcellerex XDR-50 dual-purpose bioreactor from GE Healthcare were compared with a 75-L stainless-steel fermentation vessel (DCI-Biolafitte).
Biostat CultiBag RM systems are commonly used for mammalian-cell growth. With the Xcellerex XDR-50 dual-purpose bioreactor, we used a bag designed for low– to medium–cell-density microbial cultures. We assessed performance of the three single-use systems on the basis of operability, process performance and scalability, and product quality. Operability was assessed by comparing differences such as operational parameters, utility requirements, and time and resources required by each system. We used our company’s platform process for pDNA production to compare process performance among the systems and assess pDNA quality. To compare growth profiles, plasmid productivity, and quality achieved by each system, we used E. coli DH1 expressing model plasmids that averaged 6.0 kb. And we assessed scalability by comparing results obtained for respective single-use systems against trended batch data using systems currently in place at Cobra Biologics.
Results and Discussion
System Operability: To ensure that our company’s standard pDNA processes and procedures could be transferred successfully to the single-use technologies under evaluation, we compared them with our in-house systems to expose potential risk areas and mitigate problems before, during, and after our study. Table 1 summarizes the specific parameters we assessed before and after the trial, outlining the main equipment and operational changes and results obtained for all systems tested. Use of an ambr250 modular system completely removed the requirement of vessel autoclaving and ancillaries’ preparation that were needed with a traditional reusable 7-L bench-top Applikon vessel. The modular bioreactor vessels came presterilized (by gamma irradiation) with precalibrated pH and dissolved oxygen (DO) probes inserted. Elimination of vessel and ancillary equipment cleaning and set-up before and after operations reduced time and resources (labor and materials) required for operating the modular system by 75% from those needed with the 7-L bench-top vessel.
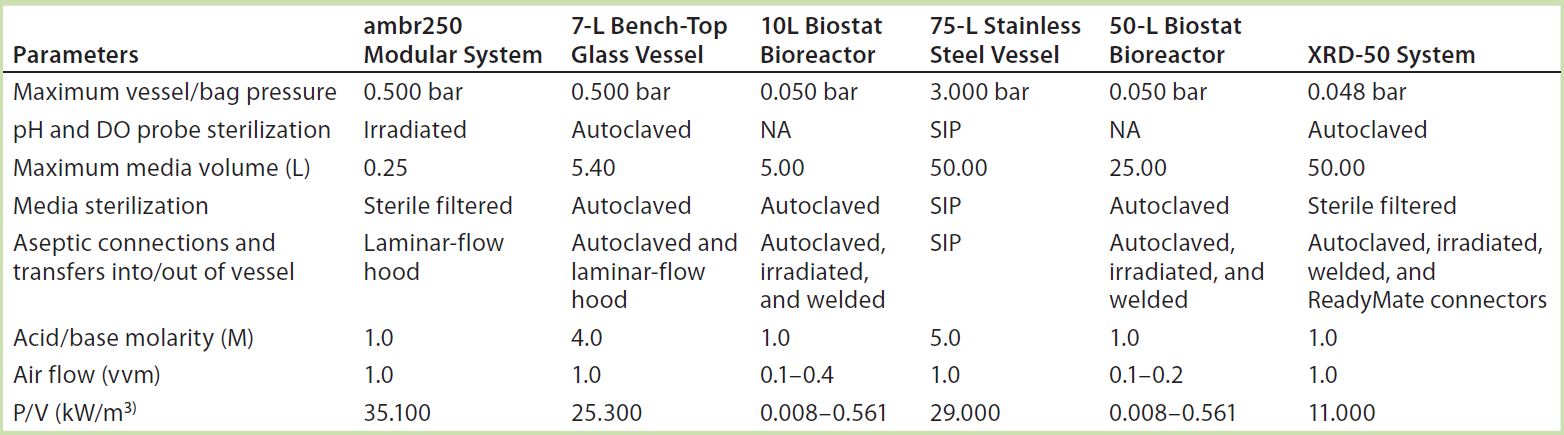
Table 1a: Summary of parameters assessed before operation of systems tested; standard Biostat CultiBag bags were modified accordingly (P/V values for Biostat bioreactors are from Reference 12; calculated P/V values are otherwise based on maximum agitation rates); NA = not applicable.

Table 1b: Summary of parameters assessed after operation of systems tested; standard Biostat CultiBag bags were modified accordingly.
Both rocking systems (10 L and 50 L) and the XDR-50 stirred-tank system eliminated the need for time-consuming and resource-intensive vessel clean-in-place (CIP) and sterilize-in-place (SIP) activities required for the 75-L Biolafitte stainless steel vessel. These operational set-up changes reduced time, labor, and material resources by 50% for the Biostat system and 35% for the stirred-tank system. For the CultiBag systems, an increased efficiency saving came from using optical/chemical sensor technology for pH and DO measurement. But the Xcellerex system required traditional pH and DO probes, which needed to be calibrated and autoclaved before insertion into the presterilized (irradiated) bioreactor bag. The XDR bag is not yet supplied with precalibrated and presterilized pH and DO probes. So the stirred-tank option was unable to completely eliminate the requirement to autoclave materials in preparation for a new CGMP manufacturing campaign.
We made additions to the ambr250 system under a laminar-flow hood to maintain sterility during set-up and monosepsis during operation. For the Biostat and the XDR-50 systems, we assembled and inflated sterile bioreactor bags and then pumped sterile media into them. All connections for addition assemblies (e.g., inoculum, antibiotics, feed, acid/base, and antifoam) were made either with a welder or by using ReadyMate sterile connectors from GE Healthcare.
Material properties of the plastic components limited all systems to ≤1 molar acid/base concentrations. This reduced-molarity requirement significantly increases acid/base volumes during operation. Consequently, we had to reduce initial media volumes. This made careful planning of processing volumes necessary to account for the additional acid/base quantities. Reduction of media will have a corresponding impact on final volumetric productivities and should be taken into consideration, particularly at pilot scale (50 L Biostat and XDR-50 systems). At laboratory scale (ambr250 modular system), a strictly planned sampling regime prevents significant culture-volume reductions (and resulting lower productivities).
The air flow required for plasmid DNA production in the 7-L glass and 75-L stainless steel vessels was 1 vvm, with a power/volume ratio (P/V) of 24.3–29.0 kW/m3. A minimum of 5 kW/m3 should be reached to deliver the levels of turbulence required by microbial cultures (10). The ambr250 modular system could meet the air flow (1 vvm) and agitation (35.1 kW/m3) requirement for microbial cultivation. The rocking bioreactor could deliver only a maximum air-flow rate of 0.1 vvm with reduced agitation capacity (because of the wave motion instead of an impeller agitation system). So we anticipate that injection of pure oxygen into these culture vessels would be necessary (but still insufficient) to meet microbial culture respiratory demands.
To increase the maximum air flow rate of the rocking system, the supplier changed the bag design to allow for continuous air and oxygen feeding through the headspace. The outlet gas filter area was increased to accommodate greater air/O2 flow rates until maximum bag pressure was reached. That increase in outlet filter area improved the maximum air flow to 0.2–0.4 vvm. Furthermore, 100% oxygen was supplied to the 10-L and the 50-L rocking systems when an OD600nm of ~7 was reached.
Maximum air-flow rates delivered by the stirred-tank system coincided with those of our 75-L stainless steel bioreactor (1 vvm) although the single-use system uses an alternative mixing technology. With its lower P/V (11.0 kW/m3) than that of our current systems, we anticipated that the XDR-50 also would require a supply of air enriched with oxygen to meet microbial culture oxygen demands. As expected, the culture required addition of ≤20% oxygen into the air supply when the culture reached an OD600nm of 21.3. Our company will have to take into consideration the additional costs, procurement, and health and safety considerations involved with the use and supply of oxygen at the right quality standards, particularly when scaling up.
Cobra’s upstream platform for plasmid DNA production requires two separate temperature shifts during the fermentation process. With the 75-L stainless steel vessel, it takes 15–20 minutes to reach final set-point temperatures during both temperature shifts. We expected both the Biostat and XDR-50 systems to encounter operational problems with control and duration of such temperature shifts because plastic material is poorly conductive. But the temperature was raised during induction and maintained throughout fermentation, and both single-use systems also took 15–20 minutes to reach their set-point temperatures. Both were also able to maintain an operating temperature within desired specifications (±1 °C).
Before harvest, the culture must be cooled down. The 50-L Biostat system had no cooling capability, so we harvested that culture directly into a cooled, sterile bag. The XDR-50 system took ~60 minutes to cool to the desired temperature. Those solutions will require analysis for routine operations. For both options, the effects on resulting pDNA quality needed to be thoroughly assessed.
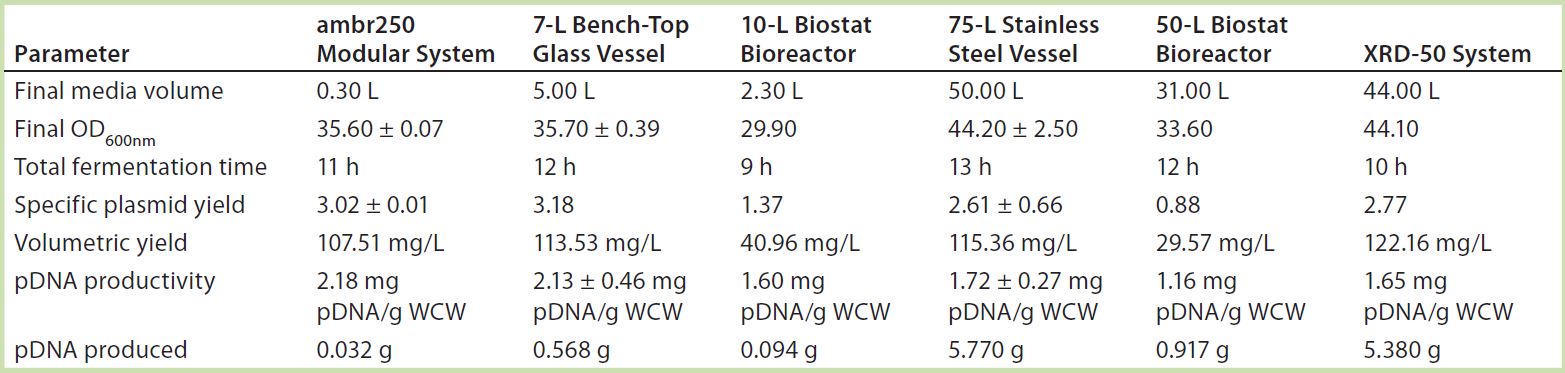
Table 2: Summary of results for plasmid DNA production in Escherichia coli DH1 in different systems and at different scales; data shown used E. coli DH1 expressing a model plasmid of 6.0-kb average size.
Process Performance and Scalability: Table 2 summarizes conditions and results that maximized plasmid DNA levels in E. coli in different single-use and traditional systems at different scales. The data come from E. coli DH1 fermentations expressing model plasmids of 6.0-kb average size.
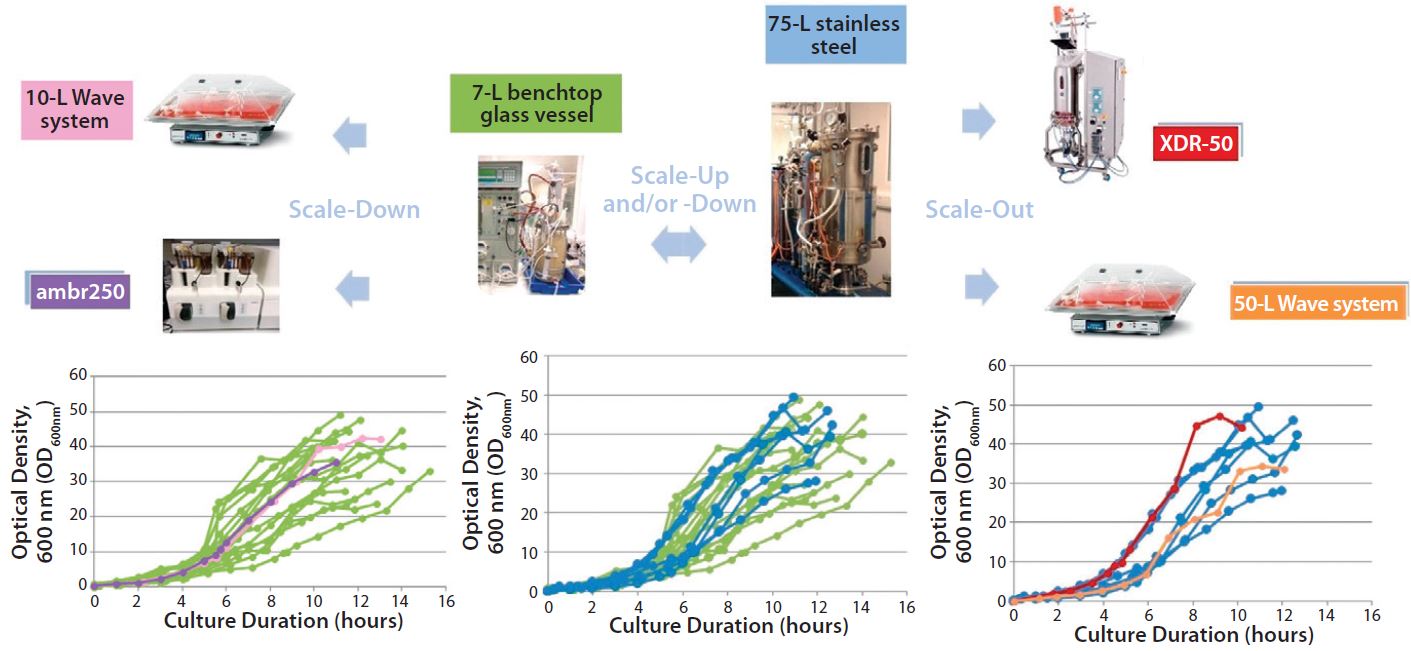
Figure 2: Scalability assessment of single-use systems tested against batch data from traditional 7-L bench-top glass and 75-L stainless steel vessels; graphs show growth profiles of E. coli DH1 represented as optical density at 600 nm (OD600nm) producing a range of different plasmid DNA sizes (2.5–12 kb) in five different systems at different scales.
Similar E. coli DH1 culture densities were achieved with the 7-L bench-top vessel, the small-scale ambr250 modular bioreactor, and the single-use 10-L and 50-L Biostat systems (OD600nm of 34–36). Culture densities attained in these systems were lower than those achieved with the 75-L scale stainless steel fermentor and the XDR-50 system (OD600nm ~44). The 7-L fermentor and ambr250 modular bioreactor were harvested about an hour earlier than all other systems, which may explain their lower final cell densities. The 10-L Biostat system reached a final OD600nm of ~42 (Figure 2), with maximum SPY levels reached at an OD600nm of 30 (Table 2).
Overall, the final culture densities reached by all systems fell within trend for those historically observed with the production platform across the 7-L and 75-L scales (Figure 2). Differences in growth did not directly correlate with SPY obtained for the systems tested. The 7-L bench-top fermentor, ambr250 modular system, 75-L pilot-scale vessel, and XDR-50 bioreactor obtained relatively similar SPY values (~3 mg pDNA/L OD600nm). Only the XDR-50 system presented a clear improvement over the Biostat system, which produced three times less pDNA. All systems presented an acceptable level of pDNA productivity (2 mg pDNA/g WCW), so we expected no additional burden on the subsequent lysis or downstream purification steps.
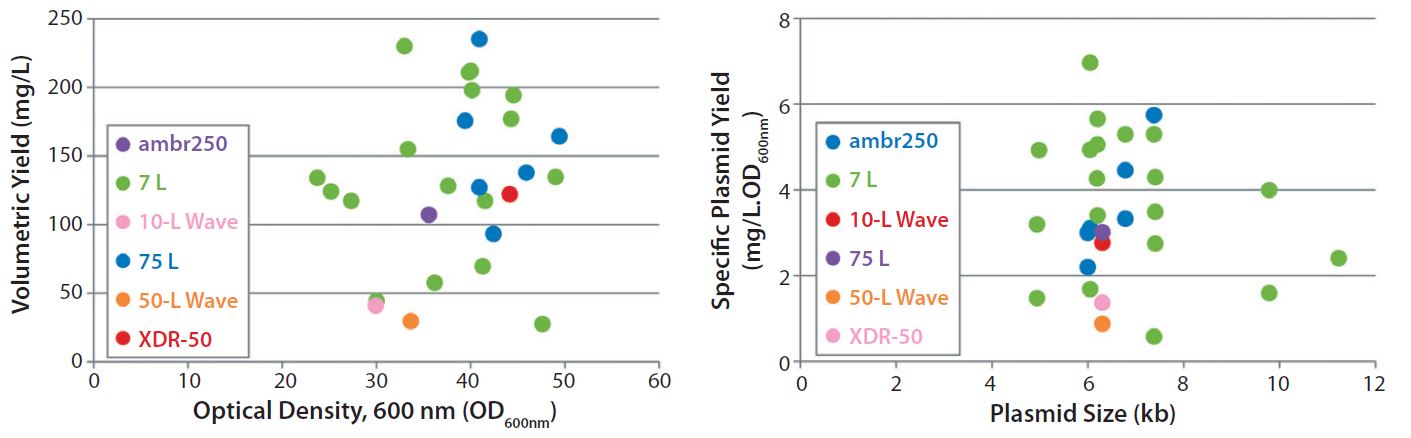
Figure 3: Scalability assessment of single-use systems tested against batch data from traditional 5-L bench-top and 50-L stainless steel vessels; graphs show (left) volumetric yields against growth (OD600nm) and (right) specific plasmid yields (SPY) for a broad range of different plasmid DNA sizes (4.9–11.2 kb) produced by E. coli DH1 using five different systems at different scales.
Figures 2–3 present a scalability assessment of the single-use systems compared with historic data ranges from a number of batches producing different pDNA products. Those data ranges came from 7-L bench-top fermentors and a 75-L stainless steel vessel. Overlapping batch data relative to the E. coli DH1 growth (Figure 2) and pDNA productivity (Figure 3) from the single-use systems show that they are directly scalable. In addition, we validated their applicability to a broad range of different plasmid sizes and types.
Note that these systems were tested as a part of a technology trial. In all instances, only a very limited number of runs (n = 1 or 2) were (able to be) conducted. Further evaluation is therefore required to appropriately justify potential reasons for specific improvements or reductions of E. coli growth and pDNA productivities. Our work shows how an initial evaluation of novel technologies would be conducted and should aid in decision-making regarding suitable technologies, with the aim of a future implementation.
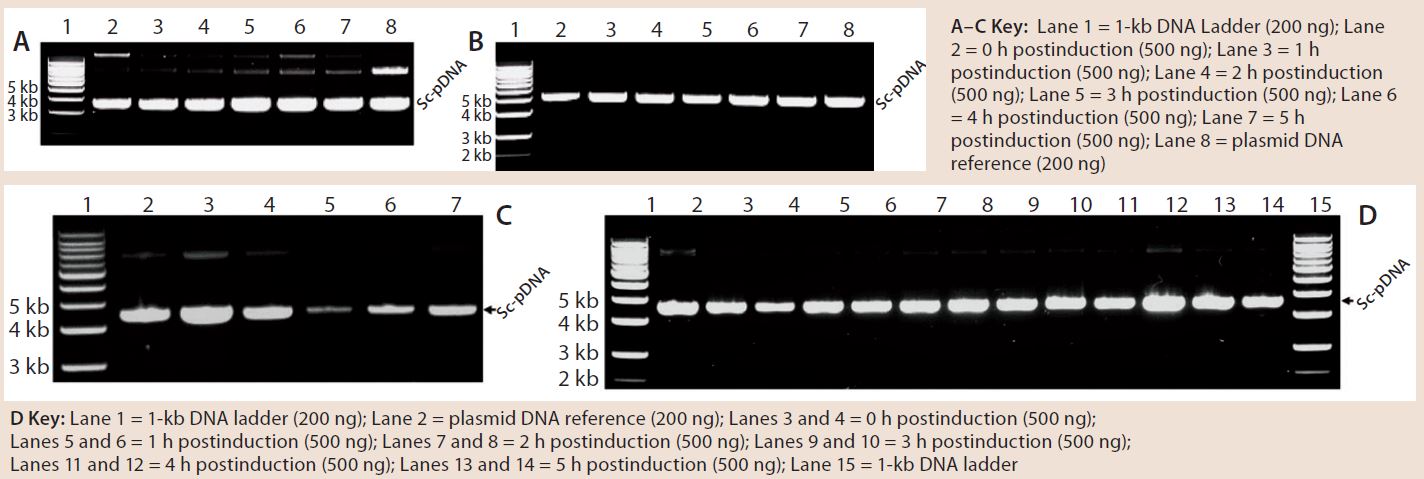
Figure 4: 0.8% agarose gel electrophoresis (AGE) analysis of a model plasmid (average 6.0 kb) produced throughout the culture of E. coli strain DH1postinduction, using different systems and scales of operation; each well was loaded with 200 ng or 500 ng of plasmid.
Quality and Purity of pDNA: Figure 4 shows some agarose gel electrophoresis (AGE) gels we used to assess pDNA quality throughout the fermentation process with different systems. Clearly, the quality of pDNA generated was consistent across all systems/scales, with no signs of gross structural or segregational instability when compared with pDNA reference material (not all data shown). As was the case for pDNA productivity, all systems generated material of acceptable supercoiled pDNA levels according to the AGE results.
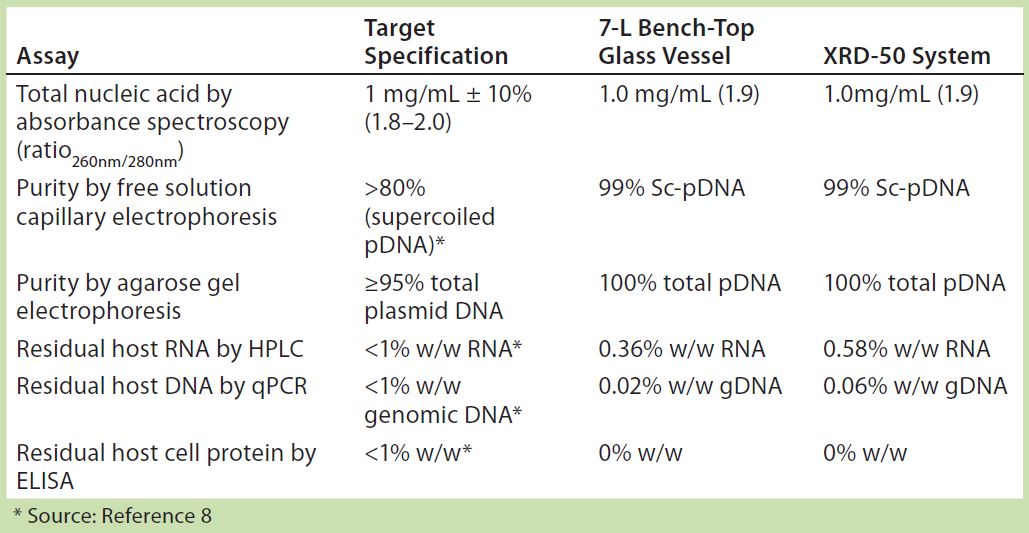
Table 3: Summary of assay results for purified plasmid DNA obtained from E. coli DH1 expressing a model plasmid of 6.0-kb average size generated in different systems — a 7-L bench-top stirred-tank glass bioreactor vessel and an Xcellerex XDR-50 dual-purpose single-use bioreactor system (GE Healthcare).
To further assess the quality of pDNA produced during these system evaluations, we selected cell paste from the XDR-50 system for subsequent purification and side-by-side comparison with material generated from our 7-L scale bench-top glass bioreactor. We purified the pDNA generated using Cobra’s in-house purification platform and subjected the purified material to a full range of analytical testing (Table 3). Results show that the purified material generated from the XDR-50 system was directly comparable to that purified from the 7-L bench-top system, meeting the critical quality attributes desired for large-scale CGMP-grade plasmid (8). These results provided confirmation that the pDNA material generated by single-use systems was consistent for purification by Cobra’s in-house platform.
Encouraging Comparability
Here we present a simple approach for preliminary evaluation of novel single-use technologies in production of plasmid DNA. Our evaluation demonstrated that the single-use ambr250 modular system, Biostat CultiBag RM bioreactor, and Xcellerex XDR-50 dual-purpose bioreactor all presented viable options for pDNA production using E. coli DH1 cultures with our company’s platform process. All provided culture growth and pDNA productivity and quality that were comparable to results achieved with in-house traditional systems.
We found the single-use ambr250 modular system and 10-L Biostat system to be suitable scale-down tools for high-throughput process development and optimization activities. The Biostat CultiBag RM 50 and Xcellerex XDR-50 dual-purpose bioreactors were more suitable for production of high-quality pDNA and CGMP material for clinical trials, respectively. All single-use systems significantly reduced our set-up and processing times (35–75%), thus increasing cost savings and flexibility to meet future capacity requirements.
This data-driven exercise has provided our company with the confidence to support single-use technology adoption and implementation of these systems. Further development and optimization are now required to integrate these technologies within our existing pDNA production platform and fine-tune associated operational process parameters to production of particular plasmid sizes and types for particular applications with each specific system.
Acknowledgments
The authors acknowledge GE Healthcare and Sartorius Stedim Biotech AG for providing all equipment used in this study.
References
1 Hitchcock AG, et al. Scale-Up of a Plasmid DNA Purification Process. BioProcess Int. 8(11) 2010: 46–54.
2 Cranenburgh R. Escherichia coli Strains that Allow Antibiotic-Free Plasmid Selection and Maintenance By Repressor Titration. Nucl. Acids Res. 29, 2001: E26.
3 Williams SG, Cranenburgh RM, Weiss AME. Repressor Titration: A Novel System for Selection and Stable Maintenance of Recombinant Plasmids. Nucl. Acids Res. 26(9) 1998: 2120–2124.
4 Cranenburgh RM, Lewis KS, Hanak JAJ. Effect of Plasmid Copy Number and lac Operator Sequence on Antibiotic-Free Plasmid Selection By Operator-Repressor Titration in Escherichia coli. J. Mol. Microbiol. Biotechnol. 7, 2004: 197–203.
5 EMA/CAT/80183/2014 (draft). Guideline on the Quality, Non-clinical, and Clinical Aspects of Gene Therapy Medicinal Products. European Medicines Agency: London, UK, 23 March 2015.
6 Subramanian G, et al. Strategies for Plasmid DNA Production in Escherichia coli. Ch1. Subramanian G, Ed. Biopharmaceutical Production Technology, Volume 2. John Wiley and Sons: Hoboken, NJ, 20 August 2012.
7 Tejeda-Mansir A, Montesinos RM. Upstream Processing of Plasmid DNA for Vaccine and Gene Therapy Applications. Rec. Pat. Biotechnol. 2, 2008: 156–172.
8 CBER. Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Modifications. US Food and Drug Administration: Rockville, MD, November 2007.
9 Hernandez OR, et al. Chapter Three: Scalable Technology to Produce Pharmaceutical Grade Plasmid DNA for Gene Therapy. Gene Therapy: Developments and Future Perspectives. Kang C, Ed. InTech Europe: Rijeka, Croatia, 2011.
10 Martínez MD, et al. Scale-Up of Fed- Batch Culture to Produce Plasmid DNA in Escherichia coli. BioPharm Int. 25(12) 2012.
11 Eibl R, Werner D. Bag Bioreactor Based on Wave-Induced Motion: Characteristics and Applications. Adv. Biochem. Engin./Biotechnol. 115, 2009: 55–87.