 by John Otrompke, with Cheryl Scott and S. Anne Montgomery
by John Otrompke, with Cheryl Scott and S. Anne Montgomery
When the United States Food and Drug Administration (FDA) approved the country’s first ever biosimilar on 6 March 2015, it had been a long time coming. After all, the European Union had approved the first biosimilar in 2006, and a number of others have followed in Europe since then. Still, the approval of biosimilar filgrastim, a recombinant colony-stimulating factor used to offset the complications of chemotherapy, was a welcome step forward for the biosimilars segment of the biopharmaceutical industry.
Amgen had sold the reference product in the United States since 1991 and enjoyed more than US$1 billion in sales in 2014. The biosimilar approval was considered by many to be good news both for Sandoz (a Novartis company, which developed it) and for patients and their insurance companies, who could expect to enjoy savings compared with the price of the reference product (Amgen’s Neupogen).
However, this first American biosimilar approval also illustrates an important point. Amgen had introduced an improved version of filgrastim years before in 2002. PEGfilgrastim is the name the developer gave to a filgrastim protein fused with polyethylene glycol (PEG), which gave it the ability to stay in a patient’s body longer (improved bioavailability). Although the concept of a biosimilar is new in the United States, the concept of a “biobetter” is anything but.

Sandoz (a Novartis company) has been an early adopter and successful biosimilar player. SANDOZ (WWW.SANDOZ.COM)
Distinguish “-Similar” from “-Better”
That small tale, one of perhaps hundreds of scenarios unfolding as the world enters the biosimilars era, exemplifies some of the quandaries that confront companies and scientists who are active in this space. If a maker tweaks a biological agent, how much “better” must it be to be labeled a biobetter? Does a developer really want its new drug to be so different that it necessitates extensive new clinical trials? Does a biobetter have anything at all in common with a biosimilar — or are the two categories entirely dissimilar?
A biobetter is more different from an originator product than a biosimilar would be — even though a biosimilar cannot itself be an exact replica (because of the complexity of biological processes, it couldn’t possibly be), and even if its maker puts some beneficial tweaks on the original drug’s properties for the biosimilar version.
So where do we draw the line? Currently, there is little definite answer to that question, especially in the United States, because the word biobetter is not an official term of art. In this context, it is simply what a biosimilar (which is fairly well defined by federal regulation) is not (1).
“Biobetter is just one of those words people have got their teeth into without really defining what it is,” says Peter Calcott, a consultant and service provider in Berkeley, CA. “Taking a proprietary drug and creating a biobetter is simply taking what you have and making it an improved drug and therefore a new drug. There are no regulations out there to deal with biobetters per se. You use the established route for a new drug. Anything that is a biobetter or second-generation drug is an innovator drug. By contrast, biosimilars are really ‘me too’ drugs.”
Different Licensing Pathways
As a result, biosimilars and biobetters follow entirely different licensing pathways. In the United States, new biologicals are rigorously evaluated by the FDA, with some being evaluated by the Center for Drug Evaluation and Research (CDER) and others (such as blood products) evaluated by the Center for Biologics Evaluation and Research (CBER).
Either way, the process is usually lengthy and expensive, and the prices for licensed biological drugs are sometimes startlingly high. Yet such drugs are widely viewed as necessary to medical treatment. Therefore, the US Congress passed biotechnology reform along with healthcare reform when it passed the Biologics Price Competition and Innovation Act, subsequently signed into law on 23 March 2010.
Consequently, the FDA was instructed to offer an abbreviated regulatory pathway for approval to a biological product that is substantially similar to an existing product. Draft guidance documents were published in 2012–2014, and 2015 has seen three final guidances so far, with new drafts promised on labeling and other topics (2).
In Europe, by contrast, the EMA has produced a number of guidances (see the “Regulatory Guidance” box), including specific annexes for manufacturers seeking approval of particular biosimilar products such as somatropin, granulocyte colony stimulating factor, epoetin, insulin, interferon alpha, low–molecular-weight heparins, monoclonal antibodies (MAbs), follitropin-alpha, and interferon-beta (rINF beta). Currently, 16 biosimilar products are available on the European market (Table 1). Also relevant is the Q5E harmonized tripartite guideline of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), an agreement between the regulatory agencies of Europe, Japan, and the United States (3).
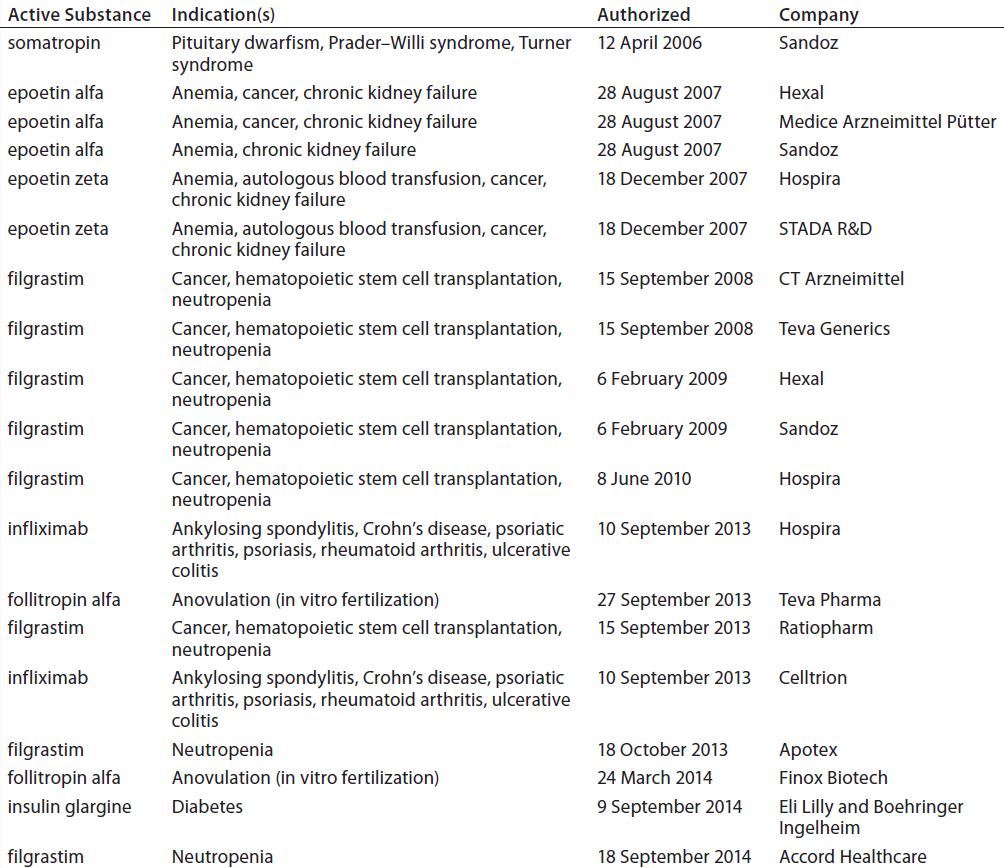
Table 1: Biosimilars on the European Union market; two others have been withdrawn from the market.
“Biosimilars in the United States are problematic,” says BPI editorial advisor Thomas Pritchett, a consultant and publisher of the BioQuality newsletter (4). “It’s not a risk I would advise a client to undertake at the moment because, although the Biologics Price Competition and Innovation Act is in place and FDA Guidance Documents have been published, the pathway to approval and how much data will be needed are far from clear. It’s much less risky, regulatorily speaking, to develop a biosimilar in Europe — not only because of the numerous precedent-setting approvals, but also because the pathway is much clearer and the guidances are product-specific. So if you can convince regulators that human clinical trials can be minimized – and if you can, for example, go from the thousands or even tens of thousands of patients needed for a big efficacy trial and maybe get it down to a few hundred patients or less for a biosimilar trial – that’s the name of the game.”
Regulatory and other Guidance
CBER/CDER. Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009. US Food and Drug Administration: Rockville, MD, February 2012.
CBER/CDER. Guidance for Industry: Formal Meetings Between the FDA and Biosimilar Biological Product Sponsors or Applicants. US Food and Drug Administration: Rockville, MD, March 2013.
CBER/CDER. Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. US Food and Drug Administration: Rockville, MD, May 2014.
CBER/CDER. Guidance for Industry: Reference Product Exclusivity for Biological Products Filed Under Section 351(a) of the PHS Act. US Food and Drug Administration: Rockville, MD, August 2014.
CBER/CDER. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. US Food and Drug Administration: Rockville, MD, April 2015.
CBER/CDER. Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product: Guidance for Industry. US Food and Drug Administration: Rockville, MD, April 2015.
CBER/CDER. Biosimilars: Questions and Answers Regarding Implementation of the Biologics Price Competition and Innovation Act of 2009 — Guidance for Industry. US Food and Drug Administration: Rockville, MD, April 2015.
CHMP/437/04, Rev 1. Committee for Medicinal Products for Human Use (CHMP) Guideline on Similar Biological Medicinal Products. European Medicines Agency: London, UK, 23 October 2014.
EMA/275542/2013. Concept Paper on the Revision of the Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins. European Medicines Agency: London, UK, 25 March 2014.
EMA/275542/2013. Concept Paper on the Revision of the Guideline on Immunogenicity Assessment of Biotechnology-Derived Therapeutic Proteins. European Medicines Agency: London, UK, 25 March 2014.
EMEA/CHMP/BMWP/42832/2005 Rev1. Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Non-Clinical and Clinical Issues. European Medicines Agency: London, UK, 18 December 2014.
EMA/CHMP/BWP/247713/2012. Guideline on Similar Biological Medicinal Products Containing Biotechnology- Derived Proteins As Active Substance: Quality Issues (Revision 1). European Medicines Agency: London, UK, 22 May 2014.
Q5E. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. US Fed. Reg. 70(125) 2005: 37861–37862.
Patents Expiring on Expensive Legacy Drugs
All the hubbub was sparked by high prices for reference biotherapeutics. But those high prices wouldn’t matter if the associated patents weren’t expiring, at least for those global regions that respect patents — or stakeholders would otherwise find a different way to express their dissatisfaction. But many original biologicals are going off patent (or soon will be), so biosimilars are expected by many experts to be an economically huge phenomenon. According to IMS Health, biologicals currently represent 18% of the overall market for drugs, or $170 billion in sales value, which is expected to rise to $250 billion by 2020 (with biosimilars making up 10% of that figure) (5). With more than 30 legacy products worth $50 billion in sales per year having lost patent protection just in the past five years, some experts predict that the market for biosimilars will have grown more than tenfold to $3.7 billion, in the same period of time (6).
The market for biosimilars was initially smaller than expected, but tepid initial uptake of such products has not slowed the stampede toward the creation of more. Although only an estimated $1.3 billion of biosimilars are sold each year worldwide at present, one analyst predicts that figure will rise precipitously to $35 billion by 2020 (5). By 2012, the FDA had received 34 pre-IND meeting requests for proposed biosimilar versions of 11 reference agents, with companies paying up to $150,000 for the chance to sit down at a meeting with the agency (7, 8). And by 2014, meeting requests had doubled.
Part of the rush might be spurred by expected potential for profit to be made. Four of the top six branded drugs that consume 43% of the Medicare Part B drug budget — Humira (adalimumab), Avastin (bevacizumab), Rituxan (rituximab), Enbrel (etanercept), Crestor (rosuvastatin), and Seretide/Advair (fluticasone/salmeterol) — are blockbuster biotherapeutics (9). (Editor’s Note: The original source erroneously refers to all six as biologics; the latter two are small molecules.) And none of those yet has generic or biosimilar alternatives available.
Notwithstanding the apparent reluctance of the FDA to approve many biosimilars, there has been little dearth of applicants willing to pay the hefty fee to sit down and discuss biosimilar applications with the agency. More than 20 preapplication meeting have been held, and the FDA has received at least nine biosimilar product licensing applications.
However, it is not just a source of profits that has people excited about biosimilars. Consumers in developed nations pay about 15–20% of the cost for the most expensive medicines themselves, with the rest paid for by insurance. Patients in developing countries such as India and Pakistan may pay as much as 55% out of pocket, instead. For some, biosimilar products may offer affordable, life-saving treatments, according to Siavash Bashiri (CEO of Xbrane Bioscience, a company based in Stockholm, Sweden), that assists with manufacturing biosimilars.
Which Pathway is Preferable?
So which would a company rather develop — a biosimilar or a biobetter? The answer to that question depends on many factors — and indeed, it may ultimately be unanswerable, for the time being at least.
Both pathways offer advantages and present drawbacks as well. If a company is developing a biobetter, then the regulatory process is much longer and much more expensive. On the other hand, if the company is making a biosimilar, then actually getting it approved is a more dicey proposition, especially in the United States. To date, biosimilar profits in the European Union have not been what was expected (5). Some experts say that if a biotherapeutic is to be marketed in the United States, its sponsor company would be better off developing a brand new, substantially improved biobetter-type drug. But one thing is for sure: If a company starts off making a biosimilar and finds that it is actually making a biobetter, the consequences could be dramatic. A company might not realize that it has developed a new drug until notified by the FDA with a denied filing!
“In less-regulated markets,” explains Xbrane’s Bashiri, “there is a big drive for cheaper medications, so these evolving markets are more keen to adopt a biosimilar approach, and they’re not keen to pay extra cost for extra efficacy that go with biobetters. But in the United States or Europe,” he counters, “the biobetter approach may be the better way to go.”
At the same time, the biosimilars business may be more attractive for manufacturers of the reference products themselves than for their would-be competitors. Sally Waterman (senior vice president of corporate development at Abzena, a biologics technology and service provider based in Cambridge, England) explains: “If you have the reference product, and three or four other companies are trying to make a biosimilar, and if you decide to do it yourself, you have all the data to do it more cheaply and more quickly, and you’re making those sales instead of your competitors.”
Some manufacturers of legacy products that are just now going off patent could create a new, “one- off” (biobetter) version of their product that is improved enough to justify a new patent. That has been going on for decades, but now it’s being considered as a means of retaining market share when a blockbuster biopharmaceutical faces competition from price-reduced biosimilars.
“For example,” says Pritchett, “if you take something in a vial and put it in a prefilled syringe and don’t change the route of administration, that’s a perfect opportunity for a biobetter.”
In some cases a company that wants to innovate on a reference product discovers the clear choice is to create a biobetter instead of a biosimilar. “One reason it can be difficult to launch a biosimilar,” says William Whitford (senior manager for cell culture at GE Healthcare Life Sciences, which makes manufacturing equipment), “is that some old monoclonal antibodies were developed ‘in the dark ages.’ Some of the old definitions were very formulaic, whereas nowadays we have so many analytics to measure a molecule’s quality attributes. Sometimes original equipment (or animals) specified are not even available. That’s a problem for some of the old molecular entities, because we didn’t even know precisely what they were at the time. For example,” he adds, “the type and extent of some posttranslational modifications weren’t even mapped in some of these original MAbs.”
However, Whitford agrees that an even more powerful reason for developing a biobetter is to compete with biosimilars manufacturers. “There are some who look at the biosimilar route and say, ‘This is so much work, we have all this expense, and in some cases it’s only a little easier than launching a new drug.’ So they say, ‘Wait a second, let’s not worry about the original molecule and just make a better new entity.’”
“Due to the different approach to biosimilars and biobetters by regulatory agencies throughout the world, there are considerable differences in the amount of analytical work required for their regulatory chemistry, manufacturing, and controls (CMC) packages, says Fiona Greer (global director for biopharma services development at SGS Life Sciences in Geneva, Switzerland). “A biobetter is treated as a novel molecule, following a normal regulatory pathway. That involves full analytical data characterization of the product as well as product- and process-related impurities, provided as Module 3 in the common technical document (CTD).
“Biosimilars, however, have their own regulatory pathways. In addition to their own CMC package, they require additional extensive comparative characterization — comparing the follow-on product side-by-side with an originator molecule. However, before that even happens, the biosimilar manufacturer must perform exhaustive structural and biological examination of the target molecule to determine its exact sequence and structure.
“That requires multiple orthogonal analytical techniques to provide data to form a quality target product profile (QTPP). This is not a ‘one-off’ event: Multiple batches should be examined over the manufacturing time frame. So the analytical burden for biosimilar manufacturers is much greater and also ‘front-loaded,’ heavier at the early stages of development than for a biobetter.”
Considering the two approaches, therefore, involves some trade-offs. Balancing the reduced need for clinical trials of biosimilars is their increased requirement for analysis using costly originator material. “SGS performs physicochemical and biological assessments of novel and biosimilar molecules,” Greer explains, “particularly focusing on posttranslational modifications such as glycosylation. We have come to understand the vast amount of data required to establish biosimilarity.” She also emphasizes the importance of an appropriate analytical strategy with the aim of designing biosimilarity into the molecule from the outset.
Case Study: Actavis Biologics
| Actavis plc is one of the world’s fastest growing pharmaceutical companies, with strong and sustainable brand franchises, a leading global generics business, and a commitment to building a leadership position in biosimilars. “The company’s $70 billion acquisition of Allergan in March 2015 creates one of the world’s top 10 pharmaceutical companies by sales revenue,” says Derek Ellison (executive director of biologics R&D, strategic partnering). Actavis has announced that it will adopt the Allergan name in the second half of 2015. The combination of Actavis and Allergan creates a global brand pharmaceutical business with leading positions in key therapeutic categories and specialization in eye care, neurosciences/CNS, and medical aesthetics/dermatology/plastic surgery, as well as a portfolio of world-renowned brands including the BOTOX injection (onabotulinumtoxin A). “Historically Actavis has focused on biosimilars in its biologics pipeline, but with the combination with Allergan, we also have branded biologics for the first time,” says Ellison, who himself joined the company when a biotechnology contract manufacturer he cofounded (Eden Biodesign) was purchased in 2010 by Actavis, which itself was previously named Watson Pharmaceuticals.“Our biosimilar product pipeline currently consists of four oncology products being developed in collaboration with Amgen,” says Ellison. “These include biosimilar versions of Herceptin, Avastin, Rituxan/MabThera, and Erbitux products representing some of the fastest-growing pharmaceuticals in the world, with peak innovator sales topping $20 billion.” Avastin is on the World Health Organization’s list of essential medicines. |
Case Study: Merck and the “Patent Pirate” Strategy
| Several years ago, Merck & Co. was working on a biosimilar version of Amgen’s blockbuster rheumatoid arthritis drug, Enbrel (etanercept). The company had paid $720 million to license a version of it from Korean company Hanwha through 2024. But only a few months later, Amgen disclosed a “submarine” process patent, and Merck abandoned the project (1). Such a patent has its issuance and publication intentionally delayed by the applicant for up to several years. This is possible with a patent system in which patent applications are not published and terms are measured from grant dates rather than priority/filing dates. US patent applications filed before November 2000 were not published and remained secret until they were granted. That allowed them to “stay under water” for long periods until they “emerged” and surprised the relevant market. Companies using such a strategy are sometimes referred to as “patent pirates.”Thus, “there are many potential surprises for biosimilar companies that are not mindful about IP issues,” points out said ChromaCon’s Bavand, “especially about originator process patents.”Late last year, Merck received another IP-related setback — this time from the other side, when a US district court judge invalidated patents held by Cubist Pharmaceuticals, which the company was in the process of buying. (Cubist manufactures Cubicin, an antibiotic used for skin infections.) But Merck planned to complete the transaction anyway (2).
References 2 Pierson R. Merck Says Still Plans to Buy Cubist, Despite Patent Setback. Reuters 9 December 2014. |
Old History with New Molecules, Little American Experience with Similars . . .
Although biobetters have been around for decades, the first biosimilar was approved in the United States only this year. Most of the regulated world’s experience in biosimilars has been in the European Union, where some 25 are licensed, but the phenomenon is still young enough that some consultants advise their clients to go slow. It may occasionally happen that a company working on a biosimilar stumbles accidentally upon a biobetter. However, as a general rule of thumb, biobetters and biosimilars are two different animals.
As an example, Calcott discussed TNF-binding drugs. “A drug I was involved with was Enbrel (Amgen’s etanercept), which was the second biologic in the marketplace after Remicade (Janssen Biotech’s infliximab) to treat arthritis. Then there came another drug, Humira (Abbott’s adalimumab). Those three molecules are different: They bind tissue necrosis factor, but they do it in different ways. Yet they’re not biobetters. They’re each developed individually, not copying the other. So they are each proprietary drugs. They each have their good points, but they are not biobetters because you cannot consider them developed one from the other.
In December 2014, Cadila Healthcare launched the first biosimilar adalimumab, which is going for $200/vial in India — where it doesn’t have to meet as stringent requirements as biosimilars do in the West (10). That’s 80% less than the Humira price. In 2012, the Abbott drug had worldwide sales of $9.3 billion, with $4.3 billion in just the United States. The Humira patent expires in the United States next year.
“Biosimilars are a new concept,” Calcott points out, “whereas biobetters have been in the development world much longer. For instance, a second-generation drug usually was improved over the first generation. Perhaps it was an improved formulation so it could be stored at room temperature rather than at 2–8 °C, perhaps it stayed in the body longer so required fewer injections, perhaps it had an altered structure with improved efficacy. That would be a biobetter or follow-on drug and would come from the innovative company. Biosimilars can come from anyone.”
“An example is a urine-derived gonadotrophin, follicle stimulating hormone (FSH),” says Michael Bavand (chairman and CEO of ChromaCon AG, a manufacturer of chromatographic processing equipment based in Zürich, Switzerland). “Serono had a first-generation product called Pergonal, consisting of a mixture of gonadotropin products. The company succeeded in purifying and relaunching it as a purer product called Metrodin, having essentially only one active principle: FSH. The product was successful because it enabled a higher take-home-baby rate than Pergonal.
“A few years later, Serono launched a follow- on product of Metrodin: Metrodin HP, which was highly pure and caused fewer reactions at the injection site. That product sold with a significant mark-up price. Again a few years later, Serono introduced the fourth-generation product, Gonal-F, a recombinant FSH that was marketed as a premium product and provided consistent quality without the imminent danger of product shortages of the drug substance due to failed product batches.
“Serono had a segmented market, offering a variety of different prices. Every 5–10 years, it would come out with a new product that was better than the previous ones. The company has been out there for 40 years or even longer now,” explains Bavand, who notes that the price for the product had doubled in some of its formulations.
Case Study: A Somatotropin Biobetter
| “There’s a hormone called somatotropin,” says consultant Calcott, “which is responsible for human growth, and people with dwarfism actually lack that protein, which is made in the pituitary gland. What companies used to do was extract the protein from the pituitary glands of cadavers.” When injected into people with growth failure, it would induce them to grow. Those who lack natural growth hormone include people with with chronic kidney failure, Noonan syndrome, Turner syndrome, Prader-Willi syndrome, and short stature at birth with no catch-up growth, among others. “It needs to be given when you are quite young,” Calcott explains.An example of a biobetter was developed in the late 1970s and 1980s, when the appearance of certain viruses (e.g., HIV and hepatitis C) brought up safety issues in relation to the use of human materials for therapeutic purposes. “In addition,” Calcott points out, “the natural sources of human somatropin (HST) were very limited in supply. So we had two drivers for a biobetter: safety and supply.”Because of the potential safety issues with cadaver- derived versions of the protein, Genentech (now Roche) developed a recombinant version that solved the supply problems as well.“ In this case, the recombinant version was a biobetter of the human- derived version. And it still required a submission and clinical trials. “A number of versions are now being prepared as potential biosimilars in Europe as Nutropin is going off-patent.” |
. . . but an Increasingly Long History with Biosimilars in the Developing World
“Actually, we’ve had the equivalent of biosimilars all over the world for years,” says Pritchett. “Many countries have approved and manufactured their own versions of biopharmaceuticals rather than importing them from a US or European manufacturer. Because of the complexity of the drug substance, these are not identical and so are essentially biosimilars.”
But BPI editorial advisor Adriana Manzi (managing director of Atheln, Inc. in San Diego) cautions, “Biosimilars are by definition compared with a reference product. Many products made in different countries are ‘copies’ of innovator products but were never compared with the reference drug and so can be called ‘me-too’ products instead.”
“To develop a biosimilar is a $100 million regulatory pathway in Europe or the United States, but in less-developed countries it is $20 million to develop a biosimilar,” says Bashiri of CMO Xbrane. “There are fewer patients in the phase 3 trial and not the same kind of documentation requirements in the ACTD” (11). That’s the common technical dossier used in the Association of Southeast Asian Nations (ASEAN) economic community.
Rather than making biobetters, Xbrane is focusing on the creation of biosimilars, he says. “We are focusing more on lowering the cost of production, and we have other clients interested in other biosimilars, such as other kinds of insulin.” Xbrane has been in existence for six years and initially focused on vaccine development, he says. Then the company split into two, with the division that focused on vaccines now a separate company. “We have grown from two to six employees,” he says about the remaining biosimilar group, “and in 2014 we moved to a bigger laboratory close to Stockholm University.”
This is an important distinction, however: The word biosimilar is usually reserved for products that are developed to be equivalent to a marketed product on the US, European, and/or Japanese markets (6). Demonstrating biosimilarity for countries such as India and China does not require the same rigorous comparability exercises. Therefore, biologics sold as “equivalent” for those markets have not necessarily been shown to be biosimilar as the FDA or EMA would understand it. Often, the phrase nonoriginal biologics is used for such products.
Illustrating the point is the fact that India has approved more than 60 “similar biologic” products for its market. By contrast, the more stringent requirements of ICH-aligned countries have limited their approvals so far: e.g., Australia with eight (Table 2), Japan with seven (Table 3), and Canada with three (Table 4). Meanwhile, China just finalized its own biosimilar rules (based on those in the United States and Europe) in March of 2015.
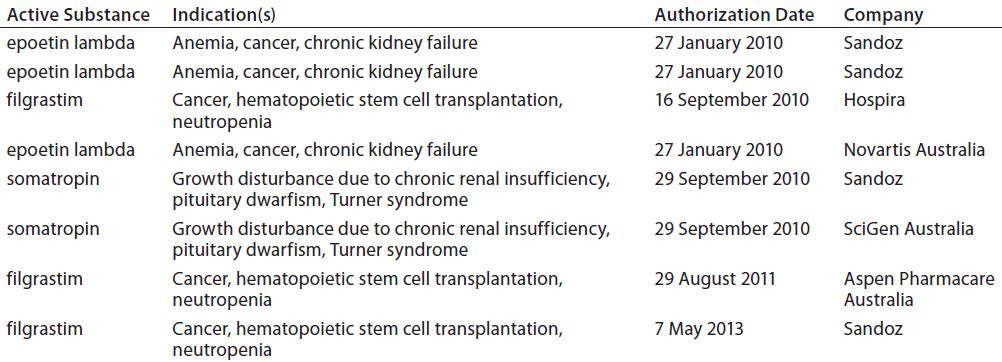
Table 2: Biosimilars on the Australian market
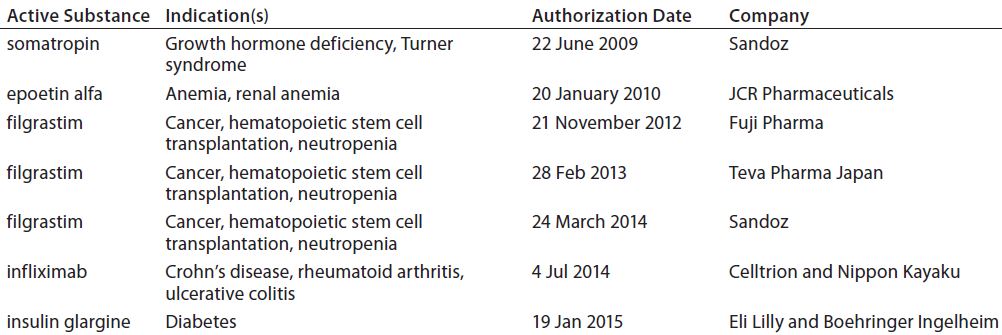
Table 3: Biosimilars on the Japanese market
Table 4: Biosimilars on the Canadian market
Manzi asserts that for some complex biosimilars, the cost of development may reach over $200 million. The more complicated analytical characterization is needed, the higher that cost.
Is the Process the Product?
So we come back to the question: What is a biobetter, and what is a biosimilar? In this new world, the conventional philosophical wisdom of biopharmaceutical development (“product = process”) may be insufficiently enlightening. As is well-known, it is impossible for one version of the same biological agent to be 100% identical to another — that is simply the nature of the beast (12). If a company makes a similar agent by a different process, especially one using modern technology that did not exist when the reference product was coined, then that company cannot expect the result to be identical. Biology involves far too many variables to keep track of. Even when an originator makes a process improvement — e.g., using advanced technology — it typically faces a gauntlet of comparability testing to prove that the resulting product is “the same.” And depending on the type of change, such testing may include a new clinical trial.
Most biosimilars are and will be made with very different processes from those used to manufacture the reference products. This is out of necessity (the mother of invention) because makers of reference products often hold process patents on the techniques used to create their drugs — another means of extending the life of the drug patent. If the FDA required every biosimilar agent to be made identically to the original substance it references, then there might be no biosimilars at all. And that would contravene the spirit of the Biologics Price Competition and Innovation Act. Instead, the agency requires substantial similarity, which is measured by the totality of evidence.
“Successfully making a biosimilar that is similar enough to satisfy regulators is a very complex process,” says Pritchett, “because it’s not just one problem, but many problems. You could, and do, make a slightly different molecule with every batch. For me, it’s also a measurement problem. Say we all decided to measure your conference room, and we all used the same tape, but we didn’t share the measurement with each other. Would we all get same measurement?”
And Manzi points out, “Or we all measure the room but do not share which method we used to do so. The measurements would differ because the methods are different.”
In the course of developing a biosimilar product, a company often has to come up with new processes — and therein lies a growth industry. New bioprocess technologies developed over the past couple decades can offer new and more efficient ways of making a biotherapeutic. And that could be crucial to the success of such products because biosimilars are all about the concept of cost reduction.
“Usually what happens is that if want to make process changes, some you can make just by yourself, but a good number of them require a submission to the agency — not a new application, but simply an amendment to the application,” says Calcott. “Most drugs out there have gone through postapproval changes; however, for biotech products often a lot more data are required to prove that a change is benign. Now, for a change from interferon A to the PEG interferon A, that requires a new submission with clinical data. That constitutes a second-generation, follow-on, or a biobetter drug.”
Case Study: Chromacon’s Technology
| Another novel manufacturing technology is twin- column countercurrent chromatography. “With ChromaCon’s processes operated using Contichrom CUBE equipment, you can circumvent infringing other process patents and also achieve significant reductions in cost of goods (CoG),” says ChromaCon’s chairman and CEO, Michael Bavand.Like many other companies, ChromaCon has been investing in innovative technology to facilitate biosimilar development. For example, two of its process patents were recently granted in Japan.“We have a new technology to enrich particular protein isoforms to create biosimilar protein isoform mixtures that are practically identical to an originator product in isoform profiles,” says Bavand. “ We can also use this technology for efficient isolation of product- related impurities, which is very important for preclinical regulatory assessments,” Bavand explains. |
Intellectual Property Complications and Opportunities
| In a 2011 panel discussion online (1), Paul A. Calvo (Sterne, Kessler, Goldstein & Fox PLLC) described the intellectual property (IP) aspect of the biosimilar–biobetter question: “For both types of products, a clear understanding of the intellectual property surrounding the target molecules is of utmost importance. This not only includes patents that cover the compositions and methods of their use, but also those that cover second-generation patents on, e.g., formulations, dosage forms, and upstream/downstream processing.” In the same discussion, Gregory Bell (Charles River Associates) explained, “The upcoming patent cliffs facing manufacturers of blockbuster biologics are daunting. . . . Biosimilars and biobetters will change the way major life-sciences companies do business — for instance, allocating resources to high-science R&D, aligning their production and commercialization assets, seeking partnership opportunities, developing and executing risk mitigation strategies, and shifting their corporate culture and focus away from brands and toward therapeutic area portfolios and service solutions. . . . The industry is likely to become much more dynamic and strategic than we’ve seen with small-molecule generics.”Finally, consultant Rajeshwari Hariharan forecasted, “Biobetters are the next wave.” She said that biosimilar manufacturers looking to strengthen their long-term strategy will acquire or develop technologies for improved biologics to help them compete against second-generation innovator biologics. “Many believe that biobetters, with their improved characteristics, are a more exciting and favorable proposition and will offer medical advantages along with a better price.”Reference 1 Competitive Strategies in Life Sciences: Biobetters Versus Biosimilars. Financier Worldwide November 2011; www.financierworldwide.com/ competitive-strategies-in-life-sciences- biobetters-versus-biosimilars/#. VUqZavC7Jp4. |
New Processes Can Contribute to Branded Biologicals
Because biosimilar developers are expected to take advantage of innovations in manufacturing, both to reduce costs and get around process patents, business is booming for contract manufacturing organizations (CMOs) and process innovators. New technologies may even lead to product improvements. But the hope is that end products will be similar enough to the originals to warrant use of an accelerated licensure pathway.
An expansion of manufacturing facilities also reflects the technological ramp-up. It is estimated that new techniques required to make biosimilars will necessitate expansion of manufacturing facilities worldwide. More than 700 biosimilars or biobetters are in development (13). If they make it through the pipeline, as many as 25% more new facilities will be required globally. And they will need to be technologically advanced and require highly efficient bioprocessing.
Supporting that growth has been GE Healthcare’s life sciences business, which in recent years acquired a number of companies that manufacture technology for use in developing biosimilars as well as novel biotherapeutics. Those acquisitions include HyClone cell culture media and sera (part of a $1.2 billion purchase of strategic assets from Thermo Fisher Scientific), and Wave bioreactors (single-use equipment that uses a rocking motion to facilitate cell suspension and gas exchange in the culture process), as well as Xcellerex (maker of larger-scale, stirred-tank single-use bioreactors).
“GE has acquired five or six key companies in the past five years that contribute toward our total, start-to-finish offering,” says GE’s Whitford. “We were known for expertise in downstream and wanted to build upstream capabilities so we could provide a fully integrated approach. That helps customers continuously optimize every stage of their manufacturing process, with the potential to reduce costs, increase yields, and reduce time to market.”
Increased acceptance and sophistication of single-use technologies are providing a major economic advantage to companies developing follow-on biologics (as well as those working on new innovator products). “Cost reduction is one of the advantages of a single-use bioreactor,” says Whitford. “Both facility build and equipment costs are substantially lower than for glass and steel reactor designs, and you also save because you don’t have to install all the clean-water and -steam systems to clean them.” GE estimates that capital expenditures for a single-use facility is ~30% less than that of a stainless-steel equivalent.
“Looking at operations,” Whitford goes on, “there is a reduction in personnel requirements and, because of the reduced services requirement, a reduction in overall quality systems maintenance. In the event of a process or product change, you can unplug a single-use reactor from the wall and move it down the hallway. However,” he cautioned, “you do have to invest in the product-contact bags for each use.”
Reflecting the surge, the number of single-use units sold has doubled in some markets just over the past two years, Whitford says, and the sale of some GE cell culture products in particular has been increasing by double digits every year for the past several years. The company also provides extensive applications data, training, and bridging manufacturing services to customers who are in the process of building a new facility, updating an old one, or otherwise installing equipment.
Another company that has found value in GE’s products is BioOutsource, a contract bioanalytical and biosafety testing laboratory based in Glasgow, Scotland and specializing in biologics and biosimilar MAbs. It was recently acquired by Sartorius Stedim Biotech Group, a leading international supplier for the biopharmaceutical industries. That acquisition is intended to further develop the group’s service portfolio and enable BioOutsource to invest in new adjacent services internationally.
“We’ve spent close to a million pounds worth of investment in buying new technology and new equipment and coming up with new methodologies,” says Daniel Galbraith (chief scientific officer at BioOutsource). “One of the most important ones is a Biacore surface-plasmon resonance analysis system made by GE. We’ve actually worked with some biobetters using the same assay. Anything we do for a biosimilar is directly available for somebody who wants to develop a biobetter.”
Galbraith explains that, “as a contract testing company, we verify that the copy has the same critical quality attributes as the innovator.” His CRO started up in 2007, two years after the first biosimilar was approved in Europe. And business is booming. “Staffwise, we’re approaching 100 people, having doubled in the past two years, mostly on the back of the biosimilars business we’ve seen. Our square footage by the end of this year will have grown threefold to accommodate new laboratory space.” The company currently works for 84 customers, he adds.
Most biosimilar developers agree that single- use technology offers them an advantage. “We invented our bioreactors and made the whole process totally disposable,” says Sarfaraz Niazi (CEO of Therapeutic Proteins International, TPI, a Chicago-based company that aims to produce biosimilars). “We have compressed and condensed everything so it can fit in one building, and we need fewer people to run the processes. If you were to use traditional deep-tank technology, you’d probably need a space 50× what we have, and you’d probably not be able to establish this company in the United States because of the high cost.” TPI plans to apply to the FDA for approval to make nine biosimilar molecules, including filgrastim, PEGfilgrastim, rituximab, and infliximab. The company also has more than 50 patents on manufacturing technology, Niazi says.
“Once you’ve proved that you can produce the same agent as a reference biological,” says Xbrane’s Bashiri, “then you need to compete with it on price. Many traditional Escherichia coli systems have an on-and-off switch to induce production of a target protein. But in our process we use a better-controlled promoter system to express the protein of interest, so we can control how much of it we’re producing.”
When it is possible, expressing biologicals with E. coli is faster than mammalian cells. “E. coli has a faster turnaround time than mammalian cells,” says Bashiri. “And the biomass is much higher in E. coli. Also, with mammalian cells, if you get a virus in production, everything has to be discarded.
“Now, you do not get posttranslational modification such as human-like glycosylation in E. coli. And for some antibodies, you need that, but not for all of them. Some examples that can be produced microbially include Genentech’s (Roche’s) ranibizumab for eye diseases such as age-related macular degeneration, and UCB’s certolizumab for Crohn’s disease. Both are approved in the European Union.” (Bashiri notes that Xbrane does not manufacture a biosimilar version of either drug.) An example of necessary glycosylation is Roche’s Gazyva (obinutuzumab) for chronic lymphocytic leukemia, which is sometimes called “the son of rituximab.” It has improved efficacy through glycoengineering.
Abzena supports other companies that are developing novel therapeutic proteins, biosimilars, and biobetters through providing services and technologies. Those include immunogenicity assessment, protein engineering, cell-line development, and PEGylation through subsidiaries Antitope Ltd. and PolyTherics Ltd.. Sally Waterman says, “We’re one of the few companies with the NS0 cell line, derived from mouse myeloma cells.” Biosimilar companies thus can access (for a fee) that originator product cell line.
“Antitope can compare the potential for a biosimilar to produce an immune response in patients with that of its originator product as part of showing that the two are similar, or at least that the biosimilar is not worse (more immunogenic). This in vitro assay is the only one that we are aware of that has been shown to correlate with the reported incidence of antidrug antibodies (ADAs) in patients,” Waterman says. “It can also be used to confirm whether an antibody engineered to have less potential to produce immunogenicity is indeed less immunogenic. Such an antibody or protein could be considered a biobetter.”
Antitope has produced two biobetter antibodies with reduced potential to induce immunogenicity in patients. They are humanized versions of alemtuzumab (currently marketed by Genzyme, a Sanofi company, as Lemtrada for multiple sclerosis) and ipilimumab (marked as Bristol-Myers Squibb’s Yervoy for melanoma). PolyTherics has PEGylated a range of proteins to produce longer-acting versions, including interferon beta (marketed in different forms by Biogen Idec, Merck Serono, and Pfizer, with a biosimilar from CinnaGen) and octreotide (Sandostatin from Novartis). “We have data in preclinical models showing that they’re effective, so producing biobetters is something we could do for our customers,” Waterman adds. (However, she points out, Abzena does not develop or own products.)
If, however, you are developing a new technique, intending to produce a biosimilar, and find that you are producing a biobetter instead, bear in mind that it is subject to an entirely different regulatory pathway, and you should adjust your plans accordingly.
Different Paths to the Same Goal
The ultimate goal is always to bring life-saving medicines to as many patients who need them as possible at the most agreeable price, no matter who the payer will be. But as always with biotherapeutics, technical details cannot be ignored. Although the maze of intellectual property issues surrounding biotechnology is far beyond the scope of this discussion, for example, it’s worth noting that many time-sensitive and demanding legal issues relating to biosimilars and biobetters are yet to be resolved around the world. Periods of exclusivity, protected trade secrets, and confidential commercial data are defined differently by jurisdiction, as are information-sharing pathways and available infringement actions. And other international regulatory differences put marketing questions at issue from the very start of product development — and throughout the drug life cycle.
References
1 CBER/CDER. Draft Guidance for Industry: Clinical Pharmacological Data to Support a Demonstration of Biosimilarity to a Reference Product. US Food and Drug Administration: Rockville, MD, May 2014; www.fda.gov/downloads/drugs/ guidancecomplianceregulatoryinformation/guidances/ ucm397017.pdf.
2 Gaffney A. FDA to Publish Biosimilars Labeling, Generic Opioids Guidance Later This Year. Regulatory Focus 27 February 2015.
3 Molzon J, et al. The Value and Benefits of the International Conference on Harmonisation to Drug Regulatory Authorities: Advancing Harmonization for Better Public Health. Clin. Pharmacol. Therapeut. 89(4) 2011: 503–512.
4 BioBetters Face Off Against BioSimilars. BioQuality March 2012: 1.
5 Rickwood S, Iervolino A. Shaping the Biosimilars Opportunity: A Global Perspective on the Evolving Biosimilars Landscape. IMS Health: London, UK, December 2011.
6 Rickwood S, Di Biase S. Searching for Terra Firma in the Biosimilars and Non-Original Biologics Market: Insights for the Coming Decade of Change. IMS Health: Danbury, CT, 2013.
7 Sherman RE (CDER). Biosimilar Biological Products. FDA Biosimilar Guidance Webinar, 12 February 2012.
8 CBER/CDER. PDUFA V: Information Technology/ Informatics Plan FY 2013 – FY 2017. US Food and Drug Administration: Rockville, MD, September 2014; www.fda. gov/downloads/ForIndustry/UserFees/ PrescriptionDrugUserFee/UCM416711.pdf.
9 So AD, Katz SL. Biotech Boondoggle. New York Times 7 March 2010; www.nytimes.com/2010/03/08/opinion/08so. html?_r=0.
10 Cadila’s Biosimilar at 20% of Humira Price. TotalBiopharma 9 December 2014; www.totalbiopharma. com/2014/12/09/zydus-cadila-first-biosimilar.
11 The ASEAN Common Technical Dossier (ACTD) for the Registration of Pharmaceuticals for Human Use — Part I: Administrative Data and Product Information. ASEAN Drug Regulatory Authority. September 2002.
12 Biosimilar Product Development Re-Emphasizes Importance of HCP Analysis. BioQuality BioSimilar Special Edition 2012: 3.
13 Langer ES. 11th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production. BioPlan Associates, Inc.: Rockville, MD, April 2014.
John Otrompke is a medical journalist who also specializes in conference coverage; john_otrompke@ yahoo.com. S. Anne Montgomery is cofounder and editor in chief, and Cheryl Scott is cofounder and senior technical editor of BioProcess International.
Defining a New Class of Medicine
by Ronald A. Rader and Donnie Elizabeth Gillespie
Biosimilars are a new class of biopharmaceuticals that require showing substantial biosimilarity (lack of significant differences) in structure, activity and clinical performance, enough to extrapolate safety and efficacy, relative to a prior- approved “reference” product (1). In many respects, such as basic molecular structure, biosimilars involve much the “same” active agent as their reference comparators. But many bioprocessing- dependent aspects, such as tertiary structure and glycosylation, will unavoidably be different.
Biobetters are much the same active agent as their comparators but novel enough (sufficiently better or different) to be considered new. For example, a protein may be modified chemically (e.g., through molecular fusion), physically (e.g., liposome-encapsulated), or in its formulation (e.g., an inhaled version of an injectable product such as insulin). As new products, these require full testing to support market approval (including large, costly, lengthy phase 3 safety and efficacy clinical trials). Developing biosimilars with only comparative testing thus is simpler, faster, and less expensive.
No matter what type of product they are working on, biopharmaceutical developers must evaluate their manufacturing strategies in terms of productivity and efficiency. In our 12th annual survey report, nearly 20% of such companies indicate that efficiency in production is the single most critical factor in their manufacturing strategy (2).
Comparing Biosimilars and Biobetters
The healthy biopharmaceutical development pipeline includes nearly 700 biosimilars (about 500 of them targeted to major markets) and some 475 biobetters, all targeted to major markets (3). So a large number of these products will be entering world markets in coming years, perhaps a dozen or more competing with each other and their reference products. With biosimilars, biobetters, and reference products all competing against each other, uncertainty remains about how US and world markets will evolve with this mix.
The Biosimilars Situation: Biosimilars involve bioprocessing-related risks that do not affect biobetters. These active agents and finished products need to be proven to be (bio)similar to their reference products. Even if bioprocess details and specifications have been disclosed (rarely the case), it can be difficult to closely replicate another company’s biopharmaceutical product. However, as illustrated by the number of biosimilars already marketed in the European Union, that has not been a major barrier to entry.
The Biobetters Situation: Perceived as innovative, unique, and improved, biobetters have inherent marketing advantages over biosimilars, which risk being viewed as low-end copies like generic drugs. On the other hand, biosimilars will have lower prices and approvals citing them as just as good as their reference products, which should facilitate their marketing. However, biobetters generally are priced higher than biosimilars and even their reference products (if they are actually better). So they offer more secure pricing and profits, with payers in the United States and other developed countries willing to pay a premium for improved products.
Market Outlook
Biosimilars’ market evolution remains uncertain. European uptake has been anemic, worldwide sales of biosimilars remain low (<$1 billion), and the US market is only just beginning to open. Profits and price discounts relative to reference products remain uncertain. Although 25–30% discounts predominate in Europe, the US market is likely to involve higher discounts and more competition. Already, some companies are reportedly proposing up to 50% discounts.
Multiple biosimilar (and biobetter) versions of nearly every successful reference product can be expected to enter the market. Thus, any single biosimilar will claim only a small portion of the total market (among competing similar products). For example, one biosimilar may capture ≥10% of a given market: ≥$100 million/year with a blockbuster reference product. By contrast, with biobetters being perceived as new and unique, they have a better chance of carving out new markets from this same product mix — and maybe even replacing the reference product. But biobetters that are not perceived to provide sufficient improvements risk failure on the market.
With development costs of $30–$130 million, biosimilars are less expensive to bring to market than originator and biosimilar products. Biobetters generally cost an additional $100 million or more to develop, thanks primarily to large phase 3 trials. Biosimilars take less time as well, with shorter trials and no need for original research.
Finally, in terms of patent-dispute risk, biosimilars and biobetters seem to be comparable. Both require expiration of reference-product patents and involve similar patent-infringement suits by reference-product sponsors. Biobetter developers, however, have the opportunity of patenting novel aspects of their products.
References
1 Rader RA. An Analysis of the US Biosimilars Development Pipeline and Likely Market Evolution. BioProcess Int. 11(6) 2013: S16–S23.
2 Langer ES. 12th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production. BioPlan Associates, Inc.: Rockville, MD, April 2015.
3 Rader RA. BIOPHARMA: Biosimilars/Biobetters Pipeline Database; www.biosimilarspipeline.com.
Corresponding author Ronald A. Rader is a senior research director, and Donnie Elizabeth Gillespie is a senior research projects manager at BioPlan Associates, Inc., 2275 Research Blvd., Suite 500, Rockville, MD 20850; 1-301-921-5979; rrader@ bioplanassociates.com.
From the Online Archives at www.bioprocessintl.com
Rader RA. What Is a Generic Biopharmaceutical? Biogeneric? Follow-On Protein? Biosimilar? Follow-On Biologic? BioProcess Int. 5(3) 2007: 28–38; 5(5) 2007: 20–28.
Medina C, Heilman CJ. Biosimilars and the Changing Environment of Regulation and Reimbursement. BioProcess Int. 5(9) 2007: 22–28.
Lundblad R. Throwing a Flag at Biosimilars: Risky Plays Leave Regulators to Decide. BioProcess Int. 6(3) 2008: 88.
Aagaard AW, Purdy S, Philpott S. Review, Approval, and Marketing of Biosimilars in the United States. BioProcess Int. 8(11) 2010: 12–20; 9(1) 2011: 14–19.
Bonilla JW, Beaver N. The New US Biosimilar Legislation, One Year Later. BioProcess Int. 9(5) 2011: 22–30.
Rader RA. Nomenclature of New Biosimilars Will Be Highly Controversial. BioProcess Int. 9(6) 2011: 26–33. Konski A. Generic Biologics. BioProcess Int. 9(8) 2011.
Rios M. Biosimilars in Development Current Views on Manufacturing, Analytics, and Commercialization. BioProcess Int. 9(9) 2011: 33–40.
Rios M. Clinical Development of Biosimilars. BioProcess Int. 9(11) 2011: 64. Whitford WG. Single-Use Technology Supports Follow-On Biologics. BioProcess Int. 10(5) 2012: 20–30.
Weinstein V. Looking at the Recent FDA Biosimilar Guidelines: Immunogenicity Concerns and Extension to Other Classes of Drugs. BioProcess Int. 10(6) 2012: 10–14.
Haxthausen N. Biopharmaceuticals On Demand Free from Traditional Constraints, China Reinvents Biomanufacturing. BioProcess Int. 10(11) 2012: 46–48.
Girard FC. Process Optimization of Biosimilars Production Using NMR Profiling. BioProcess Int. 11(1) 2013: 52–56.
Perkins M. Tunable Half-Life Technology: Redefining the Rules of Drug Dosing Frequency? BioProcess Int. 11(3) 2013: 60–63.
Emerton D. Deal-Making in the Biosimilars Market: Increasing Investment for Decreasing Return? BioProcess Int. 11(3) 2013: 70–72.
Di Cesare S, et al. High-Yield Production of PASylated Human Growth Hormone Using Secretory E. coli Technology. BioProcess Int. 11(4) 2013: 30–38.
Detmers F, Mueller F, Rohde J. Increasing Purity and Yield in Biosimilar Production: Taking Protein Purification to the Next Level. BioProcess Int. 11(6) 2013: S36–S41.
Maggio ET. Biosimilars, Oxidative Damage, and Unwanted Immunogenicity: A Review. BioProcess Int. 11(6) 2013: S28–S34.
Hulse J, Cox C. In Vitro Functional Testing Methods for Monoclonal Antibody Biosimilars. BioProcess Int. 11(6) 2013: S24–S27, S41.
Rader RA. An Analysis of the US Biosimilars Development Pipeline and Likely Market Evolution. BioProcess Int. 11(6) 2013: S16–S23.
Emerton D. Profitability in the Biosimilars Market: Can You Translate Scientific Excellence into a Healthy Commercial Return? BioProcess Int. 11(6) 2013: S6–S14, S23.
Gupta D, Prashanth GN, Lodha S. A CMO Perspective on Quality Challenges for Biopharmaceuticals. BioProcess Int. 11(9) 2013: 20–26.
Gagnon P. Emerging Challenges to Protein A: Chromatin-Directed Clarification Enables New Purification Options. BioProcess Int. 11(9) 2013: 44–52.
Ridgway A, et al. Biosimilar Products: Scientific Principles, Challenges, and Opportunities. BioProcess Int. 11(10) 2013. Rios M. A Global Joint Venture Strategy for Biosimilars Development. BioProcess Int. 11(11) 2013: 28–33.
Humphrey R. Evolving Biologics Demand Spurs New Facility Needs. BioProcess Int. 12(2) 2014: 72. Galbraith D. Biosimilars Awaken CROs. BioProcess Int. 12(6) 2014: S24–S27.
Wang X, Li Q, Davies M. Higher- Order Structure Comparability: Case Studies of Biosimilar Monoclonal Antibodies. BioProcess Int. 12(6) 2014: 32–37.
Chin K. Global Biotech Expansion Demands Drive Local Manufacturing Needs. BioProcess Int. 12(7) 2014: 24.
Niazi S. Process Innovation Leads to More Affordable Biosimilars. BioProcess Int. 12(7) 2014: 63.
Linderholm A, Chamow SM. Immunoglobulin Fc-Fusion Proteins Part 2: Therapeutic Uses and Clinical Development. BioProcess Int. 12(10) 2014: 2–7.
Osmane M, Brady J. Guidance Is Lacking in the European Biosimilar Regulatory Framework: Considering the Dynamic of Quality Profiles in Development. BioProcess Int. 12(10) 2014: 10–18.
Scott C. Unwanted Immunogenicity: From Risk Assessment to Risk Management. BioProcess Int. 12(10) 2014: insert.
Lange I, Chhatre S, Zoro B. Reducing Timelines in Early Process Development: Using a Multiparametric Clone-Selection and Feed-Optimization Strategy. BioProcess Int. 12(10) 2014: 34–37.
Langer ES. The US Biosimilars Future Is Hard to Predict: Global Health Policy, Innovation, Prices, and Profits Are All at Stake. BioProcess Int. 12(11) 2014: 30-33.
Remington KM. Fundamental Strategies for Viral Clearance – Part 1: Exploring the Regulatory Implications. BioProcess Int. 13(1) 2014: 10–16.
Dhanasekharan K, et al. Rapid Development and Scale-Up of Biosimilar Trastuzumab: A Case Study of Integrated Cell Line and Process Development. BioProcess Int. 13(4) 2015: 30–39.
Maybe there are more evidences should be shown to prove the fact of biosimilars and biobetters. Technology has always been proved.-Creative Biolabs
Regarding Amgen being responsible for a “submarine” U.S. patent, I don’t think that is correct. The following is from the Biosimilars/Biobetters Pipeline Database (www.biosimilarspipeline.com):
“In Nov. 2011, Amgen received U.S. patent 8,063,182 (having exclusively licensed this in 1999 from Hoffmann-La Roche) with claims including etanercept, filed in 1990 and expiring in Nov. 2028 (with Enbrel then on the market for over 30 years). Issuance of the patent had been stalled due to patent office actions (not due to stalling by Amgen, i.e., this is technically not a “submarine” patent).”
As you note, “submarine” patents are delayed and kept hidden by applicants. But here, there was no major stalling by the applicant and foreign equivalent patents were published/granted, so what’s disclosed in the U.S. patent was not hidden.
Thanks, Ron. This is what comments sections should be all about! I admit we were going from someone else’s analysis of the situation — an aspect of the business that’s somewhat obscure to science nerds like me. 😉