Biosimilars are revolutionizing the bioprocessing industry. Over the years, company CEOs have pushed different models, expanding and closing down huge amounts of internal manufacturing, research and development, and quality control facilities. Sometimes services were pushed to Asia, only to get brought back a few years afterward when a new CEO was appointed. Mergers and acquisitions further complicated those ideas. Consequently, many biopharmaceutical companies now have “gaps” in their drug development service provisions. Filling those gaps will fall on contract research organization (CROs), which now need to pick up the slack.
![BPI_A_141206SUPAR04_O_F0001g[1]](https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2014/06/BPI_A_141206SUPAR04_O_F0001g1.jpg)
JUPITER UNLIMITED (WWW.GRAPHICS.COM)
The mainstays of the CRO market have been investigational new drugs (INDs). With IND products, drug developers had over 10 years to design, modify, validate, qualify, change, refinance, fail, redevelop, and finally succeed in licensing their new “blockbusters.” Developers could choose functions to keep for themselves and what to outsource.
With biosimilars, that process is reversed: Developers start with a blockbuster and work backward in the hope of finding that they have made a good copy. All of that must be done as fast as possible following patent expiration of the innovator drug. The remit is always speed to market. So the outsourcing of many necessary tasks for successful regulatory submission becomes not only desirable, but essential. This has resulted in CROs being pushed for increased capacity and faster, more diverse services. CROs are being asked to be forward-looking and dynamic. Those companies that cannot rise to that challenge shall fall by the wayside in this sector.
Facing Advanced Technologies
Biosimilars have a relatively short history. In 2005, Sandoz finalized discussions with European regulators that outlined a pathway for the first such molecule to become approved (the US Food and Drug Administration is still in discussions on this). What became clear was that the traditional chemistry, manufacturing, and control (CMC) aspects related to the approval of a biologic would remain, but a new category would be introduced: comparability.
Developers are asked to take a complicated and sometimes heterogeneous product such as a monoclonal antibody (MAb) and prove that their copy is indistinguishable from the innovator in all important aspects. Regulators also ask that companies apply the most relevant, up-to-date methods to the analysis of their molecules to demonstrate comparability. That sounded reasonable at first, but it became more interesting as manufacturing processes changed. The practicalities of manufacturing a biologic in 2014 provide many more options than those of 15 years ago, when many innovator drugs were developed.
Manufacturing techniques and analytical technologies have not stood still in the intervening years. For example, Herceptin (trastuzumab, from Genentech) was approved in 2002, long before the widespread use of single-use bioreactors, animal-free growth media, and new mass spectroscopy technologies — and much before the industry understood the interactions of MAbs with many parts of the immune system. Manufacturers today can manufacture such biologics at reduced costs — more efficiently and safely than ever before — and with more in-depth drug characterization.
Comparability Challenges
As developers started to use advanced technologies to determine comparability of their biosimilars, their findings led them to pose the question, “How comparable are the batches of the innovator drug?” Simple techniques to release batches of product in 2002 revealed little variability, but today analysts can easily distinguish batches of the same product made on different geographical sites or following a
manufacturing changes. Developers are left wondering how best to describe the similarity of their molecules when innovators’ batches themselves can be different (
Figure 1).
![BPI_A_141206SUPAR04_O_F0002g[1]](https://lne.e92.mwp.accessdomain.com/wp-content/uploads/2014/06/BPI_A_141206SUPAR04_O_F0002g1.jpg)
Figure 1: Variability of Humira innovator batches
Therefore all is not as simple as first thought. To expedite the biosimilar regulatory process, developers truly need the support of experienced CROs to help them understand the differences in innovator drug products and identify comparability of their products.
The Role of CROs
Comparability Assays: Sandoz began biosimilar development with years of experience, but many developers were relatively new to biologics (perhaps having their origins in small-molecule work) and so lacked the necessary development skills. This was particularly true in the area of comparability assays, where there is now an extensive portfolio of technologies and assays to apply. One developer recently indicated that there were nearly 50 different assay types that could be involved in a Humira (adalimumab) comparability study.
The task of bringing such assay resources in-house is technically demanding, time-consuming, and costly. Even the largest pharmaceutical companies should look at outsourcing certain aspects of comparability testing. This sounds like a bonanza for the CRO industry, with large numbers of new customers and some very urgent requests. However, can traditional CROs fulfil such expectations?
The history and experience of many traditional CROs stem from an era when small molecules were king and the transition to focus on biologics was still under way. The analytics needed for small molecules were based on the chemistry of compounds with an atomic mass of <1 Da. Such technology has been around for at least 20 years and is well resourced, with many experienced CROs available. Biological or functional assays are largely not required for release of small molecules (another advantage). MAbs, on the other hand, are 150-kDa proteins with complex sugar residues and secondary and tertiary structures that must be taken into consideration. Present such molecules to a CRO more familiar with studying the 63 atoms that make up the recent small-molecule blockbuster drug (Pfizer’s sildenafil citrate), and it’s easy to see where there can be problems.
For some MAbs, sugar residues facilitate binding to certain immune-system components, which are important to a molecule’s mode of action. For other MAbs, we can remove all sugars and show no differences in activity. It is crucial to understand a molecule’s mode of action and examine the potential clinical relevance of this function.
The key is bringing together the assays needed to understand both a drug’s chemistry and biology. For example, Humira (adalimumab) is given as a treatment for rheumatoid arthritis, where it acts to remove soluble TNFα from the immune system and reduce inflammatory mediators. It is now known that this molecule (through sugar–protein interactions) activates separate immune-system mechanisms and destroys many cells that produce TNFα. Therefore, we can now describe adalimumab as having ADCC, CDC, and reverse-apoptosis mechanisms of action that were not discussed when the drug was first approved (
Figure 2).
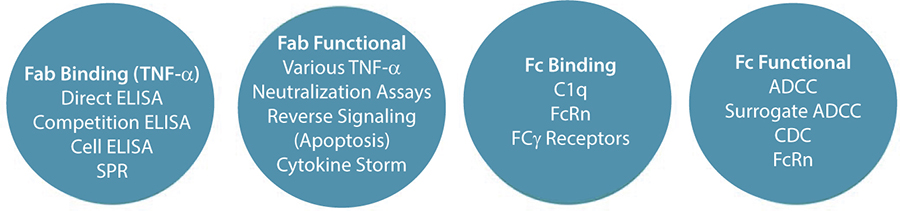
Figure 2: Methods for biological characterization for Humira and Remicade
Biosimilar Characterization: The number of techniques and assays required to adequately characterize biosimilars has presented the CRO industry with a significant challenge. Analytical assay development, qualification, and validation are time-consuming activities. Biological and functional assays also present particular challenges. Biological assays are based on cells and cellular responses, and as such they are inherently variable. As cells age in culture, they do not always perform in the same way, sometimes being less responsive to a drug during testing. Cells should be qualified in such assays to show how passage number affects response. Additional considerations include optimum number of cells to use in an assay, length of time to leave cells to settle before adding a test drug, length of response time, and best end point for an assay.
Assay Validation: Such options and parameters must be set before an assay can be validated. Validation is controlled by ICH Q2A (validation of analytical procedures). This guideline was written mainly for chemical analysis, but it also forms the basis validating biological assays. Detailing the intricacies of validating such assays is beyond the scope of this article, but a significant amount of work is required over many months to complete such work.
Some experts predict that the patents of about 15 biosimilar target molecules will expire over the next few years. If we assume that there can be as many as 40 assays required to fully characterize each of those molecules, we come to the conclusion that biosimilar developers will require close to 600 assays to support the development of their molecules.
Effective CRO Service Models
With so many assays, it is easy to see why developers would outsource such tasks to CROs. Speed to market is a crucial factor, and biosimilar manufacturers rarely have time to wait for assays to become available. But having 600 validated assays available for potential customers would stretch the capabilities and expertise of even the largest CROs.
Consequently, the biosimilar industry has (by necessity) sought out and moved away from large CROs provide a “one-stop shop.” Instead, developers are splitting their outsourced analytical services between several “boutique” service providers that specialize in particular techniques or assay types. In addition, the fee-for-service model (in which assays are developed for customers only when they sign a contract) is too slow. Such assays need to be “off-the-shelf,” for which a CRO has already invested to a sufficiently high level. Biosimilar developers need appropriate comparability assay panels for their molecules to be available on demand. With an off-the-shelf assay model, a “boutique” CRO needs to have foresight of this demand so that time is available to develop assays up-front.
The CRO–biosimilar developer model requires close partnership between a CRO and its clients. Developers share future pipeline development plans and requirements, thus shaping the service offering of their CRO partners over a period of time. In the past, CRO–client interactions were plagued by long contract negotiations involved with outsourcing complex development projects.
Off-the-Shelf Solutions
Our company’s clients are now bio-outsourcing off-the-shelf characterization assays, which are performed and reported in a matter of days. Full comparability studies are completed in a matter of weeks. Given that drugs often share targets (e.g., cancer markers), many of our biosimilar assays could be modified for new molecules. So this off-the-shelf model could be rolled out to INDs. The benefits of this model to developers are clear, but it requires a CRO to be more flexible, innovative, and involved with its client’s development plans than ever before. Biosimilars have set the alarm; it’s time for CROs to rise to the challenge.
Author Details
Daniel Galbraith is chief scientific officer at BioOutsource, 1 Technology Terrace, Todd Campus, West of Scotland Science Park, Glasgow, G20 0XA; 44-141-946-4222.
Nice article – agree this is an area for the specialists!