Many companies follow a general rule when assembling regulatory packages for presenting new biologics: Accentuate the aspects of your new biologic that mimic approved therapies. For companies working on cell-based therapies, however, that is a challenging task. The industry lacks established models, and the current European Medicines Agency (EMA) regulatory definition of a cell-based therapy is simply “an advanced therapy medicinal product” (ATMP) (see EMA guidance box).

One potential strategy is to deconstruct aspects of the therapeutic product profile (TPP). This can help you determine which components of the clinical function, upstream, and downstream processes approximate existing products and apply similar principles. As with other products, emphasis should be placed on a risk-based approach.
Defining Final Products
Many aspects of regulatory requirements governing approval of a novel biologic are linked to its intended use. A drug meant for a chronic indication in juveniles, for example, may face a higher level of scrutiny than one for a single-use treatment of a terminal disease. Linked to this scrutiny is the type of treatment (vaccine, gene therapy, immunological modifier), nature of the drug (cytokine, monoclonal antibody, virus, virus-like particle, cell, and so on), and route of inoculation. Cellular-based therapies can be categorized even further: autologous, adult pluripotent stem cells, embryonic stem cells, xenotropic cells, and /or genetically modified cells (transient and nontransient expression).
One major key to understanding the nature of a product is the development of its TPP. It requires the interaction of several company departments (e.g., discovery, PD, linical, QA /regulatory, manufacturing, and marketing), with each group being responsible for a different aspect of a product. The TPP can be as simple as a spreadsheet or as complex as a fully documented report. Figure 1 is one example of a model spreadsheet.
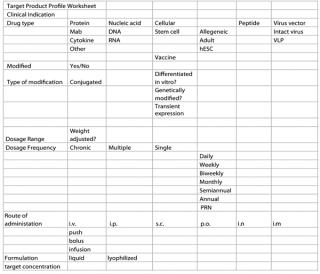
The basis of a company’s many regulatory guidance documents centers upon the definition and understanding of its final product. Many aspects of a final product are linked closely to potential clinical indications. For example, a potential patient population defines the dosage form, manufacturing requirement, and preclinical safety requirements. In turn, those requirements define the development and regulatory tasks.
Risk analysis
Incorporated in the development of a TPP should be a fully detailed risk analysis for each product attribute. Final decisions on the source of material, clinical indications, production (manufacturing) process, final formulation, and route of inoculation should all be weighed carefully against potential safety risks. Risk analysis will also provide some insight into the potential scrutiny a product will receive in a regulatory environment. Careful review of the guidelines on risk analysis (ICH Q9) and quality by design (QbD) is recommended to avoid surprises down the line (see Regulatory Guidance box).
ProDuCT origin
Related to its end use, the origin of a product can be defined in a TPP. Clinical outcomes that require replacing tissue or organs that produce critical enzymes will favor the use of pluripotent cells that may be of autologous origin. Review of various regulatory documents reveals that a key part of product characterization starts with a material ‘s origin. For existing platforms (monoclonal antibodies, recombinant molecules, vaccines), regulatory agencies place a large emphasis on the recombinant tools used to create the production platform. ICH, Points to Consider, and CHMP documents require a full understanding and description of the recombinant construct (plasmid, viral vector) and expression system used (cell line, virus).
Those documents require a full characterization of master and working stocks of cells or virus seed — a strategy that applies to cell-based therapies. ICH Q5 documents require details about the origin of a starting material. Companies must indicate whether cells are used for production of the product, whether plasmids are used, and (for cellular-derived products) the source of a product (cells) itself. Care must be taken to minimize the potential for adventitious agents derived either from the donor or the process used to derive a material. Companies must demonstrate that donors have been screened for a history of disease and disease-causing agents.
Operationally important distinctions offer unique challenges for companies producing cell-based therapies. EMA regulations (ATMP regulation article 15) concerning tissue-derived therapies, for example, require the following:
- at the tissue establishment, link between donor and donation
- at the manufacturing site, link between donation and product
- at each hospital /practice, link between product and recipient.
In addition, systems should allow full traceability from donor to recipient through anonymous coding systems. Manufacturers should establish their coding systems in a rational way — building from the coding system of a tissue establishment and designing it to facilitate tracing the donation to a product and to patients.
Regulatory Guidances
ICH
www.ich.org/products/guidelines/quality/article/quality-guidelines.html
Q5A (R1): Viral Safety evaluation of Biotechnology Products Derived from Cell lines of Human or animal origin
Q5B: Quality of Biotechnological Products: analysis of the expression Construct
Q5D: Cells used for Production of r-Dna Derived Protein Products Derivation and Characterisation of Cell Substrates Used for Production of Biotechnological/Biological Products
Q6B: Specifications — test Procedures and acceptance Criteria for Biotechnological/Biological Products
Q7: Good manufacturing Practice Guide for active Pharmaceutical ingredients
Q8 (R2): Pharmaceutical Development
Q9: Quality risk managemen
tQ11 (Draft): Development and manufacture of Drug Substances (Chemical entities and Biotechnological/Biological entities), current step 2 version dated 19 may 2011
EMAwww.emea
.europa.eu
Advanced Therapies: www.ema.europa.eu/htms/human/advanced_therapies/intro.htm
Classification: www.ema.europa.eu/htms/human/advanced_therapies/atmp_classification.htm
Certification: www.ema.europa.eu/htms/human/advanced_therapies/certification.htm
Cell Therapy: www.ema.europa.eu/htms/human/humanguidelines/multidiscipline.htm#celltherapy
Regulatory and Procedural: www.ema.europa.eu/htms/human/raguidelines/advanced_therapies.htm
Procedural Advice: www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2010/02/WC500070340.pdf
General Advanced Therapies,
EP 2.6.16: tests for extraneous agents in Viral Vaccines for Human Use
EP 5.2.3: Cell Substrates for the Production of Vaccines for Human Use
FDA
www.fda.gov/BiologicsBloodVaccines/guidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/default.htm
Guidance for the Submission of Chemistry, manufacturing, and Controls information and establishment Description for autologous Somatic Cell therapy Products Characterization and Qualification of Cell Substrates and other Biological materials Used in the Production of Viral Vaccines for infectious Disease indications
Guidance for Industry
- Eligibility Determination for Donors of Human Cells, tissues, and Cellular and tissue-Based Products
- Clinical Considerations for therapeutic Cancer Vaccines 10/2011
- Considerations for allogeneic Pancreatic islet Cell Products 9/2009
- Potency tests for Cellular and Gene therapy Products
- Cellular therapy for Cardiac Disease
- CGtP and additional requirements for manufacturers of Human Cells, tissues, and Cellular and tissue-Based Products (HCT/Ps) 1/2009
The US Food and Drug Administration (FDA) views human tissues under the current good tissue practices (CGTP). As per its guidance documents:
CGTP requirements govern the methods used in, and the facilities and controls used for, the manufacture of HCT/Ps in a way that prevents the introduction, transmission, or spread of communicable diseases by HCT/Ps (§ 1271.150(a)). Manufacture, as defined in § 1271.3(e), means, but is not limited to, any or all steps in the recovery, processing, storage, labeling, packaging or distribution of any human cell or tissue, and the screening or testing of cell or tissue donor. Under § 1271.145, you must recover, process, store, label, package, and distribute HCT/Ps, and screen and test cell and tissue donors, in a way that prevents the introduction, transmission, or spread of communicable diseases.
As with EMA regulations, CGTPs contain requirements concerning
- facilities (§1271.190(a, b))
- environmental control (§1271.195(a))
- equipment (§1271.200(a))
- supplies and reagents (§1271.210(a, b))
- recovery (§1271.215)
- processing and process controls (§1271.220)
- labeling controls (§1271.250(a, b))
- storage (§1271.260(a–d))
- receipt, predistribution shipment, and distribution of an HCT/P (§1271.265(a–d))
- donor eligibility determinations, donor screening, and donor testing (§§1271.50, 1271.75, 1271.80, and 1271.85).
That constitutes much more detail to supply for a cell than for a monoclonal antibody. It would be extremely difficult, for example, to trace back the origin of the original mouse that supplied the myeloma and the steps used in the recovery.
One major area of concern is the need to fully characterize therapeutic cells before production. Using relative markers, researchers can determine purity with respect to other cell types, but that method is limited by the sensitivity of the assays. As with novel cell types used for vaccine and recombinant molecule production, concerns about tumorigenicity and oncogencity must also be addressed.
EMA Definition of an ATMP
Advanced therapy medicinal product means any of the following medicinal products for human use:
- a gene therapy medicinal product as defined in Part IV of Annex I to Directive 2001/83/EC
- a somatic cell therapy medicinal product as defined in Part IV of Annex I to Directive 2001/83/EC
- a tissue engineered product as defined below.
Tissue engineered product means a product that contains or consists of engineered cells or tissues and is presented as having properties for, or is used in, or administered to human beings with a view to regenerating, repairing, or replacing a human tissue. A tissue engineered product may contain cells or tissues of human or animal origin, or both. The cells or tissues may be viable or nonviable. It may also contain additional substances such as cellular products, biomolecules, biomaterials, chemical substances, scaffolds, or matrices.
The Production Process
The scope and details of a manufacturing process are linked to the product’s end use as defined by a TPP. Clinical treatments that require multiple treatments or large doses for large segments of a population will result in greater challenges than “one and done” treatments for an orphan drug.
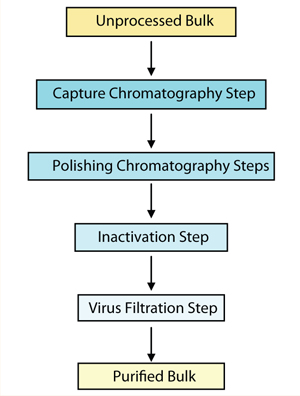
As with all biologics, the production process for a cell-based therapeutic must be fully characterized. Critical qualities such as genetic stability and limits on population doublings must be defined. In-process screening for adventitious agents is critical. But unlike for most recombinant products, the downstream process to inactivate or remove these agents usually involves very few steps. Figure 1 shows a typical purification process for a recombinant molecule. Process steps that are likely to inactivate or remove significant amounts of virus include inactivation procedures such as UV–gamma irradiation, chaotropic salts, pH inactivation, solvent or detergent treatment, and heat treatment. Adding nanofiltration and chromatography steps can provide greater viral clearance.
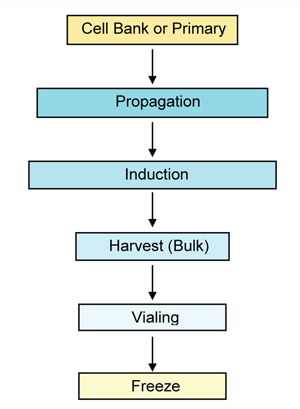
In most cases, manufacturing cell-based therapies is in essence the isolation and growth of cells followed by centrifugation, counting, resuspension in an administration buffer (with or without a cryopreservative), and vialing (Figure 2). The lack of purification steps postproduction is similar to the absence of those steps in processing some live viral vaccines and, therefore, may undergo similar regulatory restraints. Companies should review recent FDA and EMA guidance documents for vaccines (rather than the Points to Consider documents for MAbs).
Adventitious Agent Testing
Because processes for cell therapy products tend to lack downstream steps to remove or inactivate adventitious agents, emphasis must be placed on starting materials (cells, media) and containment procedures during production. Use of advanced detection techniques such as polymerase chain reaction and “ deep sequencing” are gaining interest as ways to increase assay sensitivity. Unlike myeloma cells, many stem cells and primary cells do not achieve the typical 106 cells/mL density used as the target density for current in vitro and in vivo adventitious agent assays. In addition, current live assays may be incapable of detecting many of the human agents that might be found in those types of cells.
Scale-Up and Manufacturing
Determining the final scale of production and manufacturing is related to clinical indication and market. Careful scrutiny of a target patient population before making scale decisions is highly recommended. This strategy applies to the scale required for preclinical studies, clinical trials, and commercial scale.
Targeting a population that can include pediatric patients as well as a female population of child-bearing years is likely to require a much larger scale for preclinical and clinical batches to ensure sufficient material for reproductive and studies in juvenile animals. Indications that may require multiple doses over a long period may also require a different manufacturing scale than a “one-and-done” dosing regiment because of the amount of material needed for preclinical in vivo testing.
As with all biological processes, manufacturing should be designed with validation in mind. Manufacturers must take into account population doublings during the process, control of differentiation (desirable and undesirable), and genetic stability. A robust and clinically relevant potency assay must be developed early in development. In-process assays that ensure purity (identity), safety (from adventitious agents), and efficacy (potency) developed and applied during early stages will facilitate process validation. All those process characteristics are linked to the final disposition of the product as defined by a TPP.
Regulatory Comparisons
Deciphering the regulatory requirements for a cell therapy can be a challenging task because of the unique and novel nature of each therapy. Following the examples of existing biologics can be suitable in some cases. The ability to draw the analogy to a given therapy depends heavily on a careful and thorough definition of all product attributes as described by a TPP. Using this document, manufacturers can make a careful risk analysis of each step of a development process.
Author Details
Barry Rosenblatt, PhD is president and principal consultant at SME Biotech Consulting; 1-215-694-2925; smebiotech1@comcast.net. The information provided here should be considered an overview, and consultation with regulatory agencies and expert consultants is recommended for assistance in any development program.