Live cells are being incorporated as active agents and delivery vehicles for a broad range of emerging therapeutic strategies. Successful commercialization of a cell therapy requires more than proving its safety and efficacy to regulators. Ultimately a therapy must be commercially viable, allowing enough patients to be treated with an adequate financial margin to justify investment in it as a product. “Whether the cells used are universal (allogeneic) or patient-specific (autologous), it is unlikely to be wholly one or the other that will dominate” (1). Commercial viability of conventional therapeutic products rests on economies of scale. Investment in plants, facilities, and personnel can serve a significant patient population with a defined candidature profile for a given therapy. Factories used to produce high-volume products are based around established, scalable processes with batch-control and risk-management strategies that focus on monitoring product quality. “A drug manufacturer can change the manufacturing process extensively and analyze the finished product to establish that it is the same” (2). By contrast, many emerging cell therapies will serve relatively small patient populations — with autologous therapies at the extreme, requiring a discrete batch for each patient. Furthermore, “for biologics, the product is the process. Because the finished product cannot be fully characterized in the laboratory, manufacturers must ensure product consistency, quality, and purity by ensuring that the manufacturing process remains substantially the same over time” (2). So attempts to dedicate a conventional production philosophy to producing small-batch therapies involve huge investment in cleanrooms and equipment that is often dedicated to each single small batch with “robust segregation policies and procedures” (3). Facility use is therefore poor, with extensive change-over costs when equipment is cleaned and prepared to switch to the next product batch. Processing technologies used during cell therapy clinical trials are typically based on manual laboratory methods that are not optimized or easily integrated for production. Further, although many cell therapies in development require similar processes, in reality such novel therapies often require equally novel processing technologies. Those processes also require significant manual interaction by highly trained, professional staff. Bench-level cellular therapeutic production in the preclinical stage requires skilled and highly focused personnel working for long hours in cleanroom conditions. If you watch a manual cellular process, you will see many detailed manual operations: transferring fluids from vessel to vessel, taking cell counts, controlled dilutions, and so on (Photo 1). Even if those cleanrooms are built and the staff well trained, maintaining a level of consistency and quality to satisfy regulatory requirements and process demands is challenging. Consequently, many small-batch cell therapies have manufacturing processes that are not well suited to a current good manufacturing practice (CGMP) environment. The cost of such therapies can be prohibitive, and managing scale-up to meet demand is a particularly daunting proposition (Figure 1). Photo 1

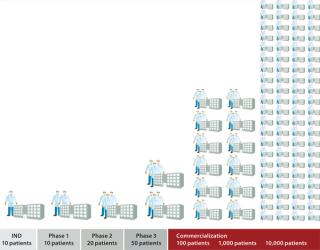
Autologous therapies appear to hold the key to controlling communicable disease transmission and addressing the need for immune suppression, both of which limit the application of allogeneic cell therapies. Despite those potential benefits, high costs normally associated with small-batch manufacturing often compromise its commercial viability and can be a major disincentive to investors and developers. Herein lies a conundrum for many companies interested in developing new cell therapies: Traditional bioprocessing factories are not appropriate for small-batch processes. Consequently, many cell therapies are developed using technologies and procedures that are inherently expensive and labor-intensive and require a high degree of skill, training, and management to minimize deviations. To develop alternative processes that are scalable will typically require significant investment over a considerable period. To prevent complications associated with postapproval process changes, a new process needs to be ready for use before clinical trials are complete — well before it is certain that the therapy will be approved for market. Several cell therapy companies have recognized the need to address this issue. Accordingly some have invested a proportion of funds allocated for clinical trials to planning, developing, and optimizing innovative, customized solutions to their manufacturing challenges. The Role of QbD and PAT There is an obvious commercial imperative for small-batch cell therapies, but process changes cannot be made without careful consideration. Biological products can be sensitive to very minor manufacturing process changes. Even relatively small process differences can significantly affect the nature of a finished product — most importantly, how it functions in a patient’s body. “When ‘the process is the product’ — as would be the case with industrialized production of stem cells — variability is the enemy and must be reduced and controlled as completely as possible” (4). Successful biological manufacturing therefore addresses scale-up, process comparability, and process characterization. Because cell therapy products cannot be fully characterized in a laboratory, manufacturers must ensure product consistency, quality, and purity by ensuring that their manufacturing processes remain substantially the same over time. So companies must tightly control the source and nature of their starting materials and establish appropriate process controls for each unique product and/or manufacturing process. Further complicating these matters is the fact that during research and early stage clinical trials, most cell therapies and associated process controls are developed and produced for a limited number of patients. They use technologies that are readily available rather than what will ultimately be needed for commercial-scale production. Typically those are manual, laboratory-scale technologies that are not optimized for specific cells or process needs. Authors from Aastrom Biosciences, Inc. (www.aast rom.com) highlighted in 2008 the criticality of batch failure for autologous therapies: “The risk that a process will fail to yield a finished cell dose meeting specifications must be extremely low, whatever the reason. For PSCT, manufacturing failure is not merely an operational inefficiency issue; instead, it directly translates into a failed patient treatment” (5). By adopting a quality by design (QbD) approach to developing cell therapy processes, we can identify and address critical failure modes and process parameters. For example, one useful measure of process risk is the number of sterile connections that must be made throughout cell therapy manufacturing. In a typical manual process, operators make multiple sterile connections in a biological safety cabinet (BSC) over several days to add reagents, collect quality control (QC) samples, and package a final product. In one relatively simple process we analyzed recently, we counted 76 sterile connections, all of which were performed manually. Each connection carries an inherent risk to the product. When patient numbers are small, such risk is theoretically manageable by skilled operators using good standard operating procedures (SOPs). But risk to products is considerable in commercial-scale production. A natural consequence of the QbD approach to redesigning such a process would be to minimize the number of manual sterile connections. In fact, when this concept of minimizing sterile connections is taken to the extreme, it implies a process without any manual sterile connections. It is therefore hardly surprising that much innovation in small-batch manufacturing for cell therapies has revolved around using presterilized closed consumables (single-use technology). Process analytical technology (PAT) has been defined as “a system for designing, analyzing, and controlling manufacturing through timely measurements (that is, during processing) of critical quality and performance attributes of raw and in-process materials and processes, with the goal of ensuring final product quality” (7). This philosophy for monitoring and controlling product quality is independent of batch size, and its implications can be profound as those batch sizes reduce. For example, consider QC for an autologous therapy. Regulators are likely to require that the same tests be performed regardless of batch size. Each QC sample must be a specific volume to suit a given analytical method. Due to economies of scale, the volume of cells used for a large batch and the subsequent cost of performing QC tests are relatively minor. However, for an autologous therapy, such identical QC samples are likely to represent a significant proportion of the total cells available — and the total cost per patient. Final-product release testing must satisfy a difficult combination of constraints. Such tests must be rapid (for products with short shelf-lives), relatively inexpensive (because they must be performed for every dose), limited in necessary sample volume (to prevent excessive loss of final product), and able to test a complex product composition. (5) So for small batches, it is even more important that we invest efforts in characterizing processes so that we understand precisely which parameters need to be monitored, controlled, and tested — including operating limits within which a predictable outcome can be assured. If we combine the need to take samples for PAT and QC with the objective to use closed consumable processing, then we have a specific small-batch requirement for which a solution must be found. How do we efficiently collect samples from the right place, at the right time, without affecting product sterility (Photo 2)? Photo 2:

Closed Systems Segregation is the comprehensive, verifiable isolation of an internal process from external contamination at all points in a processing system. All process equipment and materials must be cleaned and sanitized or sterilized between batches to prevent contamination. Cleaning and sanitization/sterilization processes also need to be validated. Batch-to-batch segregation represents a challenge to all therapeutic manufacturing processes. For small batch sizes, however, the impact of conventional cleaning and sanitizing strategies on operating costs and facility uptimes can be significant. The bioprocess industry has recently seen an emergence of presterilized, closed-consumable kits and associated equipment. These have been used for achieving product isolation in conventional cell culturing processes. As closed-consumable technology has matured and gained acceptance, the same philosophy has also been applied to several upstream and downstream processes such as closed, continuous centrifugation. KBI Biopharma Inc.’s (www.kbibiopharma.com) kSep technology is one recent example that illustrates the potential of closed processing and how scalability can be addressed. The closed, continuous centrifuge was developed to gently concentrate, capture, and/or separate cells by providing a very low-shear and nourishing environment. The kSep technology is being applied in a single-use format to take advantage of shorter cycle times, fewer validation requirements, lower processing costs, and minimal risk from cross contamination. Photo 3:

Photo 4 shows two kSep variants based on an identical core process technology but capable of significantly different throughput rates. Using them, development work can be performed at small scale with confidence: If or when larger-scale batches need to be processed, a bioequivalent solution will be available. Photo 4:

As mentioned, closed-consumable kits enable us t o address the risk of contamination during manual operations such as sterile connections. Such kits are also effective for ensuring batch-to-batch segregation. For cell therapies, each process is unique. They typically involve collection and isolation, then culture, expansion, and manipulation of cells followed by harvesting, washing, filtering, and concentrating them; formulation and product filling, storage, and transportation; and finally administration to patients. Assuming that a cell therapy process can be designed so that it remains closed throughout, implications on cost and quality can be profound. Because a closed consumable is sterile, associated risk of contamination from its environment can be considered negligible. On that basis — and provided that a consumable remains closed throughout the entire process — processing can be undertaken in lower-grade cleanrooms. It also means that different batches (provided that they too are closed) can be concurrently processed in the same room. This could significantly reduce the need to build and operate expensive cleanrooms. To realize the full potential for a cell therapy using closed-consumable processing therefore requires a “closed” solution for every manufacturing and QC step in the process, and herein lies the challenge. Although closed processing solutions now exist for some process steps, they are often not ideally suited to the manufacturing requirements of a specific cell therapy and thus are not readily integrated. Reengineering a cell therapy process to make it closed and satisfy all requirements for a specific cell therapy requires
- a comprehensive understanding of the process and its critical parameters
- thorough knowledge of the capabilities of available technologies
- identification of technology gaps and development of novel solutions
- integration of a “system” for a user-friendly, error-free process that can be quickly and reproducibly scaled to match demand (Photo 2)
- a familiarity with regulatory requirements for CGMP manufacturing.
Closed and Automated Cell Processing How It Works: Cell therapy processes including centrifugation, incubation, media addition, cell selection, cell washing, and final fill and finish can be performed within “closed” consumables (Figure 2). Machines integrate and automate the processes and replicate many qualitatively controlled manual tasks. They also provide consistent and operator-independent quality. Because processing is “closed,” automated machines producing therapies for different patients can be placed side-by-side in a processing room, whereas conventional “open” processing would require a separate sterile environment for each patient’s therapy.

Benefits delivered from an automated, closed-cell processing system would include significant reduction in the cost of therapies (typically 25–90%) and the number of operators required (typically >70%); lowered dependence on skilled labor; significant savings in capital investment through better facility use (typically 30–50%); improved quality and fewer quality events; and an ability to more rapidly scale up and scale out to match market demands. An integrated cell therapy manufacturing facility (Figure 3) comprises a central “processing room” containing several automated machines that process multiple patients and batches in parallel. Around that are situated other facilities and processes needed to support those operations including, supply, storage, and retrieval of input and output materials including reagents, disposable processing sets, patient materials, finished products, and waste; collection and processing of QC samples for data capture and process feedback; and a process recovery suite. This allows use of specialized facilities to be maximized and enables rapid scale-up of production to match demand.
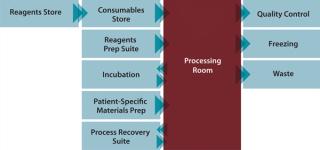
Scale Up or Scale Out? My company has been involved in developing several automated systems for both autologous and allogeneic cell therapy processes. Despite differences in the types of cells and processes required to produce these therapies, the scale-up challenges they face are remarkably consistent. Cost: Cell therapy batch sizes are often small (especially for autologous products), so applying conventional, large-batch processing strategies is a poor use of facilities. “Large GTP/GMP cleanroom facilities are impressive and glamorous, but very costly to build, and even more expensive to operate” (3). Dendreon Corporation (www.dendreon.com), which received US FDA approval for its Provenge prostate cancer treatment in 2010, specifically describes some risks to scale-up: The costs of expansion of our facilities and investment in related equipment has been and will continue to be a very significant expenditure for us during 2010 and 2011. In order to commercialize Provenge, in the event of licensure by the FDA, we will need to hire and train a significant number of employees and comply with applicable regulations for our facilities, which are extensive. In addition to the monetary costs of expansion of our manufacturing capabilities, the facilities build-out requires significant time and attention of our executive management (6). Automation: Most cell therapy processes have been developed in a laboratory. As a consequence, they are manual and labor-intensive, and they require a high degree of skill. This may be logical and manageable during research, but it would be considerably more difficult for commercial-scale production. Processing technologies typically used during clinical trials aren’t often optimized or easily integrated for cell therapy production. In a CGMP production environment where the product is the process, change is only possible under very strict controls. Changing manual processes into more efficient, automated processes after regulatory approval is often difficult, time-consuming, and expensive. In fact, we have seen several cases in which cell therapy companies are “committed” to technology that they know is suboptimal or, worse still, no longer available. A trained researcher’s mindset is forever locked into the quest for innovation and improvements as well as the drive to act on new findings — in short to experiment. Minds normally dedicated to investigating unexplored elements at the forefront of medicine would need to “switch gears” to engage in operating a production process that is reproducible, consistent, and invariably repeatable. This fundamental mismatch in mindset would be a waste of talent and training. Companies embarking on a route to scale-up, unsurprisingly, report worker dissatisfaction and significant difficulties in recruitment and retaining of staff. Risk: “The potential of cross contamination and/or loss of patient identity (chain of custody) for each batch must be maintained at a zero risk level as scale-out occurs” (1). Apart from the inherently high cost of manual cell therapy processes, it is therefore extremely difficult to imagine how you could efficiently scale up or scale out a manual process without significant deviations. Up to 50% of batch failures are attributable to operator error, and it is intuitive that the increasing complexity of an operation raises the likelihood of errors occurring (1). The primary strategy that Invetech has applied to small-batch processing is to create a new type of factory in which manufacturing processes are closed and the most complex and vulnerable process steps automated. With careful design and a real-time quality control system that detects erroneous states before process deviations are created, a manufacturing process can thus be created that achieves a commercially viable cost of goods and eliminates critical failure modes. The system developed by Invetech for Argos Therapeutics represents a previously manual process that was successfully redesigned based on the guiding philosophy of closed-consumable processing on an integrated and automated production machine. We developed an automated cellular and RNA processing system to manufacture autologous immunotherapies for treating advanced kidney cancer (metastatic renal cell carcinoma), B-cell chronic lymphocytic leukemia, and HIV infection. At the heart of the automated process are three standalone units: an RNA processing subsystem (Photo 4) that processes tumor homogenate into amplified tumor RNA, with all steps performed on a single patient sample and taking place in a closed disposable container; and two cellular units designed to perform a series of cellular and plasma processing steps to generate a final product in functionally closed disposable vessels. The system removes complex and vulnerable manual operations and replaces them with automated processing steps that can be consistently reproduced. Each unit can be validated and will perform precisely the same function no matter where in the world it is operating. This substantially eliminates the most significant contributor to variation: operators. We now have a process that can be scaled out to match demand by installing additional, automated machines in either the original facility or in others that are located closer to potential markets. By locking down the process with automation and closed consumables, we have also significantly reduced the company’s reliance on hiring and training skilled operators as well as the cost and time required to establish dedicated cleanrooms. Roadmap to Commercial-Scale Production Cell therapy companies need to invest in alternative technologies to ensure that they can have commercially viable products. However, it is also important to recognize that funds are typically precious during early stage clinical trials. Until a therapy is proven to be safe and efficacious, and a reasonably mature process exists, investment in highly customized systems should be kept to a minimum. But after phase 3 clinical trials using a specific process are complete, changes to that process can be extremely difficult to make and will require proof of bioequivalence or even additional clinical trials. Developing a customized CGMP system will typically take two to four years, depending on the complexity of the process and the time it takes to progress through development, clinical trials, and capital raising. Invetech’s recommendation is to invest a relatively small amount of effort and money at phase 1 to identify the most appropriate scale-up system and plan a development strategy that includes cost and timing (Figure 4). Another primary objective of the feasibility and planning stage is to identify core technologies that can be used for initial trials and subsequently scaled up and integrated into a CGMP commercial-scale production system.

Ideally, development of a commercial production system should commence during phase 1 or 2, depending on the development time scale and clinical trial schedule. At a minimum, the key objective should be to have a commercial-scale system available during phase 3. This ensures that those pivotal trials are conducted using a process that is equivalent to the commercial production process, which prevents a need to make expensive and time-consuming postapproval changes. Photo 6 shows one of the outputs of such a feasibility study that my company undertook for MolMed SpA before commencement of phase 3 clinical trials for its “TK” therapy. The product is an ex vivo cell therapy to enable safe haematopoietic stem cell transplants (HSCTs) from partially compatible bone-marrow donors to treat hematological malignancies, particularly high-risk acute leukemia. Photo 5:

Photo 6:

Debate will continue as to the relative merits and likelihood of success for allogeneic and autologous cell therapies. Ultimately, the overall commercial success of any such product will depend on it being safe, efficacious, cost-effective, and scalable. It also needs to be affordable and accessible to a substantial patient population. For some companies, allogeneic cell-based therapies are attractive because they reflect the current large-batch pharmaceutical investment model and have been supported on this basis. However, not all allogeneic therapies would be considered large-batch products. Many autologous therapies could provide significant therapeutic advantage. At Invetech, we believe that for the cell therapy industry to reach its full potential, it must have commercially viable small-batch processing — and ideally the ability to deal with a batch size of one (patient). Some cell therapy companies have already invested in developing innovative closed, automated systems for small-batch cell therapy manufacturing. Those systems have delivered significant cost, quality, and scale-up benefits over existing manual, cleanroom-based processing strategies. They have also provided valuable insight into the industry’s needs and demonstrated what is possible. Cell therapy no doubt is an extremely exciting and rapidly advancing field of medicine that could have a significant impact on how we treat a vast array of diseases. To reach its full potential, however, the technology will require more than old-fashioned factories. Closed and automated cell-processing systems provide an opportunity to change the paradigm and deliver these therapies at a cost that was not previously thought possible.
Author Details David James is director of life science and pharmaceutical manufacturing innovation for Invetech Pty. Ltd., 495 Blackburn Road, Mount Waverley, Victoria 3149, Australia; 61-3-9211-7742; david.james@invetech.com.au, www.invetech.com.au.
REFERENCES