Lentiviral vectors are important tools for gene transfer because of their ability to transduce a number of cell types without the need for host cells to be dividing (1, 2). As a result, investigators are using them as gene delivery vehicles in clinical applications (3,4,5,6). Although these vectors are used routinely in many research laboratories, large-scale production using current good manufacturing practice (CGMP) methods comes with a set of challenges that must be considered as more clinical trials using lentiviral vectors receive regulatory approval (7).
One important consideration in designing a CGMP-compatible process is the need to have a manufacturing protocol that produces consistent lentivirus for multiple CGMP productions. Several methods are commonly used to transfect cells with lentiviruses: e.g., lipofection, electroporation, and calcium phosphate transfection. The selected method must consistently produce high-titer virus. Although several laboratories have produced stable packaging cell lines containing certain viral genes (8,9,10,11,12,13), development of such lines is time consuming. In addition, use of stable lines does not allow the flexibility to change envelope and accessory genes or to readily alter the ratios of helper genes expressed.
PRODUCT FOCUS: GENE THERAPIES AND DNA VACCINES
PROCESS FOCUS: PRODUCTION AND DOWNSTREAM PROCESSING
WHO SHOULD READ: PRODUCT AND PROCESS DEVELOPMENT, MANUFACTURING
KEYWORDS: VIRAL VECTORS, TITER, CELL CULTURE, SCALE-UP, DISPOSABLES, GENETIC ENGINEERING, FILTRATION, YIELD
LEVEL: ADVANCED
With a mind toward generating a flexible process to produce lentivirus for a range of clinical protocols, we chose to use a four-plasmid system. Using self-inactivating (SIN) vesiculo-stomatitis virus G (VSV-G) pseudotyped lentiviral vectors to generate lentiviral particles (14), replication incompetent particles are produced by cotransfection of three helper plasmids encoding separate packaging functions as well as a separate transfer plasmid containing the vector backbone. Helper plasmids encode gag/pol and the rev-responsive element (pCgp), rev protein (pCMV-rev2), and the heterologous VSV-G envelope protein (pCMV-G) (15).
To produce lentivirus suitable for transducing a range of patient-specific cells in diverse clinical trials, conditions must be optimized to produce the highest viral titers. We focused on maximizing titer and quantity of virus produced upstream while maintaining a consistent downstream process of concentration and purification. Although many parameters involved in lentiviral transfection have been assessed by others at the research scale using T-flasks, we developed a scalable method for efficient transfection at the multiliter scale using 10-layer Corning CellStack trays. This process allows for production of large quantities of lentivirus required for phase 1–2 clinical trials (16).

Using antibiotics in production of materials for human clinical trials is undesirable. As a result, many clinical biologics are filtered as a final step in the manufacturing process. However, we and other investigators have observed that sterile filtration of lentivirus (0.2-µm pore size) decreases viral titer (17). Thus, we preferred not to invoke that sterilization method in our process. As a result, it was essential to take every precaution to ensure product sterility at all steps of the manufacturing process. To address those concerns in maintaining sterility of the lentiviral product — as well as for manufacturing staff safety — we devised a semiclosed manufacturing process that contains the virus during upstream production.
Our semiclosed system can be used as part of a modular manufacturing process involving subbatches of lentiviral supernatant. This multiple-subbatch system is particularly helpful inaddressing safety concerns that accompany production of large volumes of lentiviral material (100 L) by allowing for pooling of multiple 10-L subbatches made over several weeks. However, it is crucial to show consistent viral production across those subbatches. By optimizing upstream parameters, we developed a process that allows for extensive scale-up in a safe, sterile, and reproducible system to produce clinical-grade lentivirus. Our manufacturing process has produced several lots of lentivirus that have been or are currently being used in ex-vivo gene therapy trials.
Materials and MethodsPlasmids: For transfection of HEK 293T cells we used three lentiviral helper plasmids (kindly provided by JK Yee) produced under CGMP conditions at the Center for Biomedicine and Genetics. For each plasmid, a bacterial master cell bank (bMCB) was generated and release tested (including plasmid DNA sequencing). A single vial of the bMCB was used to produce a seed culture to inoculate either a Wave Bioreactor unit (GE Healthcare) or a Bioflo 3000 bioreactor (New Brunswick Scientific).
Harvested bacteria were lysed with a modified alkaline lysis method (18). We clarified the lysate with centrifugation and depth filtration, then ultrafiltered it 10- to 15-fold using a hollow-fiber cartridge (GE Healthcare) with a 30-kDa nominal molecular-weight cut-off (NMWC). We concentrated the resulting supernatant and passed it through a bed of USP-grade Celite diatomaceous earth from Celpure (Sigma Aldrich) to reduce endotoxin content.
We separated pDNA from bacterial RNA and other impurities using size-exclusion column chromatography with Sepharose 6 Fast Flow resin (GE Healthcare) in the presence of 2M ammonium sulfate (JT Baker). After collecting the first peak containing pDNA, we passed it through a second Celite bed to further reduce endotoxin. Column-purified material was ultrafiltered and diafiltered into Tris-EDTA buffer (Fisher Scientific), yielding purified bulk pDNA.
HEK 293T Master Cell Bank and Culture of 293T Cells: The 293T cells we used for lentiviral vector production are expanded from a master cell bank (MCB) established at our center. That bank was fully release tested according to FDA guidelines for releasing cell banks and products (19). Cells grew in culture at ~2 × 104 cells/cm2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, 1% sodium pyruvate, and 1% glutamine (Biowhittaker) supplemented with 10% Hyclone fetal bovine serum (FBS) (Thermo Fisher). We used a 37 °C incubator with 5% CO2.
Every three to four days, we trypsinized and counted the cells before reseeding them at 2 × 104 cells/cm2. After removing media from the cell culture flasks, we washed them with phosphate-buffered saline (PBS) from Mediatech and then removed that as waste. Trypsin (Irvine Scientific) was diluted 1:10 in PBS to
a final concentration of 0.05% and then added to the cell culture flasks, which were returned to the 37 °C incubator for 3–15 minutes. After cells were dislodged, we collected the cell suspension and inactivated the trypsin by adding 293T medium (roughly equal volumes of cell suspension and medium), then collected cells using centrifugation. Those cells were expanded until we had sufficient numbers for transfection.
Lentiviral Transfection for Process Optimization Studies: For cell culture condition optimization experiments, we plated 293T cells in an appropriately sized vessel at 0.5–1.5 × 105 cells/cm2 in 293T growth media (DMEM supplemented with 10% FBS, 1% sodium pyruvate, and 1% glutamine). Two days later, we transfected those cells with four lentiviral plasmids: the pCgp plasmid containing the gag/pol gene, the pCMV-Rev 2 plasmid containing the rev gene, and the pCMV-G plasmid containing the VSV-G gene — all under the guidance of cytomegalovirus (CMV) promoters — and a transfer plasmid (HIV-CMV-EGFP) in a ratio of 20:13:5:20 respectively with 0.8 µg DNA/cm2.
To make about a liter of transfection mixture (for transfecting a 10-layer tray system), we mixed DNA from three helper plasmids and the transfer plasmid with TE buffer at pH 7.9 (~50 mL). We added 2 M CaCl2 (7 mL) from Fluka (Sigma) and 2× HEPES-buffered saline (HBS) at pH 7.2 (58 mLs) in sequential order before mixing (HEPES from Roche; NaCl and Na2HPO4 from Sigma). These volumes were scaled down for smaller-scale experiments. Immediately following, we removed the medium from each vessel and replaced it with the DNA mixture in growth media with 1 L 2% FBS. The cells were returned to the 37 °C incubator with 5% CO2. Three to five hours later, we removed the transfection solution and added fresh 2% FBS-media with 6 µM sodium butyrate from Fluka (Sigma) to the Cell Factory trays. About 72 hours after transfection, virus-containing crude supernatant was collected.
CGMP Lentiviral Manufacturing Using a “Semiclosed” Manifold System: HEK 293T cells are expanded in culture and transfected in subbatches of 10 10-layer Corning CELLbind CellStack trays each. Specifically, we thaw a vial of 293T cells and place them in a Nunc T-75 flask. Those cells are passaged every three to four days into Nunc T-500 flasks and then transferred to 10-layer Nunc Cell Factory trays as expansion continues. We expand the cells until sufficient numbers are obtained to plate the number of trays required for an intended vector production and vessels needed for cell-train maintenance.
The 293T cells are plated in each 10-layer tray at a density of 1.0 × 105 cells/cm2. Two days after plating the 293T cells, we transfect the cells with four lentiviral plasmids using the calcium phosphate method. We often modify the ratio of helper plasmid DNA for each transfer plasmid to optimize the titer for an individual product, although we have found that a ratio of 20:6.3:3.2:20 (pCgp:pCMV-Rev2:pCMV-G:transfer plasmid) generally works well. The transfer DNA, which is specific to each product manufactured, is first mixed with TE buffer at pH 7.9. Then 2M CaCl and 2× HBS (pH 7.2) are added in sequential order (0.018 mL/cm2), and the solution is mixed well. We remove the existing medium from the trays before adding the transfection mixture to the 293T cells using the first of three manifolds, which allow reagents to be removed or added as part of our semiclosed system.
Manifolds connect the CellStack trays with bags containing media and reagents as well as waste bags. The culture chambers are incubated in a 37 °C incubator with 5% CO2 for three to five hours. The transfection medium is removed at the end of that incubation and replaced by growth media with serum (2%) and 6 µM sodium butyrate using a second manifold. Then the chambers are returned to their incubator. On day 3 after transfection, we collect supernatant containing secreted lentiviral vectors using a third manifold.
Directly following harvest, the crude culture supernatant is first clarified by depth filtration using either a 0.45-µm capsule filter (Sartorius) or a 0.5-µm capsule filter (Millipore) with 3,000 cm2 of surface area. We add Benzonase nuclease at 50 U/mL in the presence of 2 mM Mg for six hours at 37 °C. Clarified and Benzonase-treated viral supernatant is ultrafiltered from 10 L down to 2 L using a hollow-fiber cartridge with a 500-kDa NMWC (GE Healthcare). The viral supernatant is further ultrafiltered (UF) to ~500 mL while simultaneously diafiltering (DF) with 8 L chilled DF buffer (4 g/100 mL lactose from JT Baker in DPBS from MediaTech). While maintaining the volume at ~500 mL, we diafilter the viral supernatant with an additional 2 L of DF buffer. After UF/DF, the viral suspension is centrifuged for 16–20 hours at 6,000g. The resulting viral pellet is resuspended and then centrifuged at 1,000 rpm for one to two minutes to pellet and remove insoluble material. We then collect the supernatant and keep the concentrated viral vector material at –80 °C until all subbatches are pooled.
Transduction Unit Titer Assay: To determine viral titer efficiently and precisely, we used a lentiviral vector expressing green fluorescent protein (GFP) in our optimization studies. Each lentiviral sample was titered by transducing HT-1080 cells, and the expression of GFP was measured by fluorescence-activated cell sorting (FACS) analysis to determine the viral titer.
In brief, 5 × 104 HT-1080 cells (from ATCC) were seeded in each well of a 12-well plate using standard growth medium (DMEM supplemented with 10% FBS) and incubated for ~24 hours in a tissue culture incubator at 37 °C with 5% CO2. A single well of cells was counted to approximate the number of cells transduced in each well. Cells were transduced with various amounts of test samples in the presence of 4 µg/mL polybrene (Sigma). A previously titered viral sample of HIV-CMV-EGFP lentivirus served as a standard to control for assay variations. To quantify the number of cells transduced by the vector, we removed the HT-1080 cells from their plates ~48 hours after transduction and fixed them with 3.7% formaldehyde (Sigma). We further analyzed those fixed cells using an EPICS XL-MCL flow cytometer (Beckman Coulter) to detect GFP expression. Based on the following formula, we determined lentiviral titers:
Titer (Transduction Unit/mL) =% of GFP-positive cells × number of cells transduced ÷ volume (mL) of sample used for transduction.
We performed a p24 Titer Assay using the HIV-1 p24 ELISA kit from PerkinElmer (catalog #NEK050). Briefly, a test article is incubated in microplate wells coated with a highly specific mouse monoclonal antibody to HIV-1 p24. Captured antigen is complexed with a biotinylated polyclonal antibody to HIV-1 p24, followed by astreptavidin-HRP (horseradish peroxidase) conjugate. The resulting complex is detected after incubation with ortho-phenylenediamine-HCl (OPD), which produces a yellow hue that is directly proportional to the amount of HIV-1 p24 captured. Absorbance (490 nm) of each microplate well is determined using a microplate reader. We calculated p24 concentration based on a standard curve generated by p24 standards with known concentration.
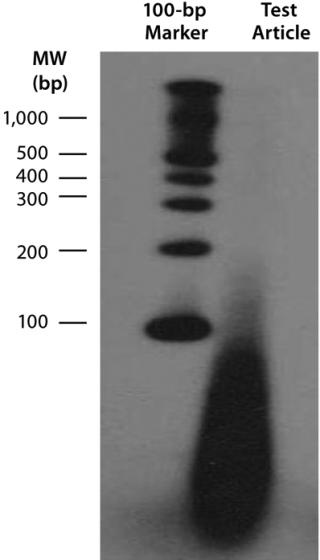
End-Labeling Assay for Determining DNA Fragment Size Distribution: We radioactively labeled nucleic acids (including residual plasmid DNA, contaminating host DNA, RNA, and any other free nucleic acids) contained in our lentiviral product by incubating it with alpha-32P-deoxyribonucleotide triphosphates (dNTPs) from MP Biomedicals and the Klenow fragment of DNA polymerase from New England Biolabs at room temperature for 30 minutes. Unincorporated nucleotides were removed using a G-50 microspin col
umn (GE Healthcare). We analyzed end-labeled DNA fragments using agarose gel electrophoresis and imaged them with X-ray film. We evaluated the size of host DNA fragments present by comparing samples with end-labeled DNA molecular-weight markers run in parallel during the electrophoresis.
Statistical Analysis: We determined statistical significance using an unpaired, two-tailed p-value Student’s t test, with a p value of <0.05 considered significant.
ResultsComparing 293 and 293T Cells: Our first step in optimizing the upstream manufacturing process required identifying a lentiviral production cell line. Master cell banks of HEK 293 and HEK 293T cells were generated and tested for sterility and mycoplasma, as well as a panel of adventitious agents according to FDA guidelines (19). Both lines have been used previously to produce lentiviral vectors with transient transfection (20, 21). We compared them for their ability to consistently produce high-titer lentivirus.
The 293 cell line (the parental cell line of 293T) was developed in 1977 as a transformed cell line by transfection of primary human embryonic kidney cells with sheared adenovirus type 5 DNA (22). The gene encoding SV40 T-antigen was later introduced into the 293 cell line to create the 293T cell line (Stanford Line, SD-3515). The latter is known to express adenovirus early region genes E1A and E1B and complement E1-deleted, replication-defective adenovirus vectors. In addition to their ability to support adenovirus vectors, these lines are recognized for favorable tissue-culture, transfection, exogenous DNA replication, gene-expression, and protein-production characteristics.
We found that lentiviral titers obtained from 293T cells were significantly better (p < 0.01) than those obtained from 293 cells (Figure 1A). In addition, we found that we could grow 293T cells for multiple passages (~15 passages) without detectable effects on the viral titers produced. As a result, a single train of 293T cells can be used for manufacturing multiple batches of lentivirus over several months without a need to continually thaw new vials of cells. That was not always the case for long-term culture of the 293 cell line, with much more variable viral titers when cells of increasing passage number were transfected (data not shown).
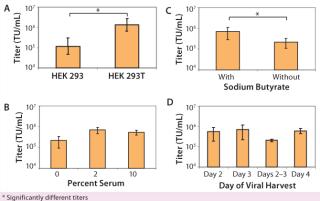
Use of Serum During Transfection: In most basic research laboratories, the 293T cell line is expanded in culture in the presence of growth media supplemented by 10% serum. In developing a CGMP-compliant manufacturing protocol with minimum animal reagents in the transfection process, we wanted to minimize impurities (such as serum proteins) from the transfection and viral production phase. Thus, we assessed the effects of adding either low serum (2%) or no serum (0%) during transfection and viral production. We fully tested our serum for the presence of adventitious agents and secured large quantities of the same lot to maintain a robust and consistent process. Adding 2% serum during transfection and viral production was beneficial to production of high-titer lentivirus (Figure 1B). Additional serum (10%) produced no higher titers. As a result, we use 2% serum in all production runs to optimize virus yield.
Use of Sodium Butyrate During Viral Production: Sodium butyrate enhances viral production during transfection (23, 24), and we determined its effect on viral titer. HEK 293T cells were transfected with the four lentiviral plasmids. About three hours after transfection, the cellular medium was removed and fresh medium (with or without 6 µM sodium butyrate) was added. Adding sodium butyrate about three hours after the plasmid was beneficial (p < 0.01) in production of high-titer lentivirus (Figure 1C).
Harvest Time: Using parameters determined above (addition of 2% serum during transfection and viral production and addition of sodium butyrate about three hours after transfection), we tested the optimal time for harvesting lentivirus after transfection. HEK 293T cells were transfected with lentivirus (day 0) in T-flasks, and viral supernatant was harvested on day 2, day 3, or day 4. By collecting lentiviral supernatant on day 2, replacing the harvested medium, and then collecting lentiviral supernatant produced during the subsequent 24 hours (“day 2–3” sample), we also tested whether multiple harvests of the same culture increased the amount of lentivirus. A single collection of viral supernatant on day 3 produced the highest average lentiviral titer (Figure 1D). In addition, multiple refeeds and harvests between 48 and 96 hours after transfection did not appear to increase overall virus yield significantly (p > 0.05). So we routinely collect virus at a single time point on day 3, which fits in well with our manufacturing process scheme. Cells can be transfected on a Friday and the cultures then harvested Monday morning.
Process Scalability: To ensure that optimized conditions would produce high-titer virus when our process was scaled up, we assessed the effect of increasing surface area on viral titer. We plated 293T cells in vessels of increasing size: from T-75 flasks to larger T-flasks (T-225) and then to trays in either one layer (636 cm2) or 10 layers (6,360 cm2). We found no statistically significant difference in titers produced among of any of the vessels tested (Figure 2A). As a result, we confirmed that a 10-layer tray was appropriate for large-scale expansion of our manufacturing process because of the associated ease of use.

Semiclosed Transfection System: We developed a closed system involving three individually designed manifolds to maintain a sterile environment for our product during transfection and harvest of crude viral supernatant. Manifolds allow media and transfection reagents to be pumped directly into trays and viral harvest to be pumped directly out into bags for further processing. We compared viral titers obtained from such a system with those obtained with an open system involving aspirator bottles. As Figure 2B shows, the difference wasnot statistically significant (p > 0.1). Considering the benefits of safety for manufacturing staff and reduced contamination risk for the product, the closed system is now routinely used for our large scale lentiviral manufacturing campaigns.
Upstream Subbatch Production of CGMP-Grade L
entivirus: We developed a process for pilot-scale production of lentivirus using the final optimized conditions. As Figure 3 indicates, 293T cells are expanded in culture and transfected in subbatches of 10 10-layer trays each. Specifically, a vial of 293T cells is thawed and placed in a T-75 flask. They are passaged every three to four days into T-500 flasks and then transferred to 10-layer trays as cell expansion continues. The cells are expanded until sufficient numbers are obtained to plate the 10-layer trays required for the intended vector production as well as to plate the number of vessels needed for cell-train maintenance. Two sub-batches of up to ten 10-layer trays each can be produced in the same week (1 L viral supernatant from each), leading to production of ~20 L lentiviral supernatant each week. Figure 3 summarizes a representative cell expansion involving multiple weeks of transfection and viral harvest.
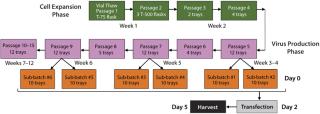
We plate 293T cells in each tray at a density of 1.0 × 105 cells/cm2. That density is well within the range (0.5–1.5 × 105 cells/cm2) that we have determined to produce high-titer virus (Figure 4). Two days after plating, cells are transfected with four lentiviral plasmids using the calcium phosphate method (25). The quantity ratio of the four plasmids (as well as the total quantity of DNA used) are often tested in an evaluation run for each transfer plasmid to optimize titer for each individual product, but we have found that 20:6.3:3.2:20 for pCgp:pCMV-Rev2:pCMV-G:transfer plasmid with a total quantity of 0.53 µg/cm2 usually works well. That ratio of plasmids is based on further optimization of previously published ratios (26) (data not shown).

DNA is first mixed with TE buffer at pH 7.9. We add 2M CaCl and 2× HBS (pH 7.2) in sequential order and mix the solution well to allow for DNA precipitation. Existing medium is removed, and the transfection mixture is added using the first of three manifolds (Figure 5A). Trays are incubated in a 37 °C incubator with 5% CO2 for three to five hours. Transfection medium is removed at the end of that period and replaced by growth media with serum (2%) and 6 µM sodium butyrate through a second manifold (Figure 5B). Then the trays are returned to the incubator. On day 3 after transfection, supernatant containing secreted lentiviral vector is collected using a third manifold (Figure 5C). That manifold allows for collection of sterile samples of crude lysate as well as end-of-production cells (EOP) required for QC product release. At the end of each week’s production, resultant crude supernatant is processed as described below.

Downstream Processing of CGMP-Grade Lentivirus: Directly following harvest, crude culture supernatant is first clarified using depth filtration with either a 0.45-µm Sartoclean capsule filter (Sartorius) or a 0.5-µm Opticap XL capsule filter (Millipore). For a 10-L batch of supernatant, we use 3,000 cm2 of surface area for the filtration process. We add Benzonase nuclease to the clarified supernatant at 50 U/mL in the presence of 2 mM Mg for six hours at 37 °C. Under those conditions, most residual nucleic acids (contaminatinghost DNA, residual plasmid DNA, RNA, and free nucleic acids) degrade into <500-bp fragments, as determined with a radioactive end-labeling assay (Figure 6). Those contaminates should be removed in subsequent process steps.
Clarified and Benzonase-treated viral supernatant is ultrafiltered from 10 L to 2 L using a 500-kDa NMWC hollow-fiber cartridge from GE Healthcare. Viral supernatant is further ultrafiltered (UF) to ~500 mL while simultaneously diafiltering (DF) with 8 L of chilled DF buffer (4 g/100 mL lactose from JT Baker in DPBS from MediaTech. While maintaining the volume at ~500 mL, we diafilter the viral supernatant with an additional 2 L of DF buffer. After UF/DF, the viral suspension is centrifuged for 16–20 hours at 6,000g. The resulting viral pellet is reconstituted in DF buffer and then centrifuged at 1,000 rpm for one to two minutes for removal of insoluble material.
Because no final sterile filtration is performed, supernatant is collected and the concentrated viral vector material kept at –80 °C while samples from each subbatch are tested for in-process sterility. Once the sterility of each subbatch is confirmed, those subbatches are rapidly thawed at 37 °C and then pooled for aliquotting into vials to complete the production. Based on several developmental runs, the overall yield of lentivirus during downstream manufacturing has been shown to be ~31% (Figure 7).

Because lentiviral vectors are important tools in gene therapy, they need to be manufactured at large scale for early phase clinical trials. To date, at least nine protocols have been approved by regulatory agencies for clinical testing using lentiviral vectors (27). Safety data from the first trials are encouraging, so we can expect the number of approved protocols to continue rising. We chose to concentrate our efforts in optimizing the upstream manufacturing process of viral production to develop a CGMP-compliant lentivirus vector manufacturing procedure that can be used to make a variety of lentiviral vectors. To determine the best conditions for scale-up, we optimized several parameters, including the choice of producer cell line, cell seedingdensities, and transfection conditions. We then determined a procedure for scalability using a semiclosed system to develop a manufacturing process that could produce lentivirus safely in a multibatch system on the 100-L scale.
We compared both HEK 293 and 293T cells for lentivirus production and found that transient transfection of 293T cells consistently produces higher-titer virus over many passages. The level of consistency seen with those cells is essential for large-scale lentiviral production, with the same vial of cells capable of producing high-titer virus over several months in culture to accommodate large manufacturing campaigns. When multiple campaigns are scheduled consecutively, overlapping cell trains can be maintained to ensure a constant supply of cells needed for virus manufacture.
We have optimized several other parameters of our upstream manufacturing process: serum concentration in the transfection and virus production medium, addition of sodium butyrate during viral production, and time of harvest. Interestingly, we found that making transfection reagents in-house provides for more consistent and higher titer viral products than does purchasing premade reagents from commercial sources we evaluated (data not shown). To ensure consistent results, our reagents are qualified before being used in a manufacturing campaign. Figure 8 summarizes our complete manufacturing process.

Using this process, we have produced and release-tested at least six lots of CGMP-grade lentivirus for multiple phase 1 clinical trials, at least one of which is complete (28). These productions have ranged from 20-L scale to >100 L (Table 1). All our viral products have been fully released for ex-vivo gene therapy trials, and Table 2 provides a sample of our release tests for one clinical lot. Final product titers were between 5 × 107 and 3 × 108 TU/mL. Final product quantities ranged from 10 mL to >500 mL based on the number of subbatches produced.
Table 1: CGMP lentivirus production using tray culture systems

Others have recently reported an ability to produce large-scale lentivirus using multilayer tray systems (29,30,31).
Table 2: Example of release tests performed on a clinical lot

One advantage of our process is an ability to produce virus in a semiclosed system with multiples of subbatches, which allows for production of >100 L viral supernatant. The semiclosed system developed for transfection and harvest of viral supernatant keeps our manufacturing staff safe and the product sterile during production. The multiple-subbatch process allows for production of large quantities of lentivirus while still ensuring the safety of our production staff by limiting the volume of lentivirus to be handled at any one time to 10 L.
This manufacturing process allows for extremely consistent viral production among subbatches of a given production (Table 3). This is crucial because the costs of manufacturing are too great to allow for a significant percentage of low-titer subbatches that could lower the overall final product yield. As can be seen in Table 2, all subbatches show extremely consistent viral production (except #4 of product lot CBG-0103.0, which had a problem in the hollow-fiber concentration step).
Table 3: Reproducibility of viral titers among subbatches

The flexible final formulation step allows individual products to be formulated in a way that best suits project-specific requirements and allows for continuous development of distinct process steps. This manufacturing process is very efficient and can be carried out using minimal staff (two operators for production of each subbatch). It provides the extensive scale-
up capacity necessary to produce CGMP-grade lentivirus, and it has been used successfully in several completed and on-going phase 1–2 ex-vivo gene therapy clinical trials.
Author Details
Corresponding author Lara J. Ausubel, PhD, is an associate research professor; Christine Hall, Anupriya Sharma, Rebecca Shakeley, Patricia Lopez, Valerie Quezada, Sylvana Couture, and Ross McMahon are manufacturing associates; Kenneth Laderman is an associate research professor; Patricia Huang is a QC associate; David Hsu is director; and Larry Couture is senior vice president of the Center for Biomedicine and Genetics at the Beckman Research Institute, City of Hope, 1500 East Duarte Road, Duarte, CA 91010; 1-626-471-7201, fax 1-626-301-8175;